Fuster Franck
Maître de Conférences à la Faculté des Sciences et Ingénierie, Sorbonne Université.

Manual
wfn file manipulations
For some specific appliacations the wfn file must be modified because GAUSSIAN contains a bug in the case of ROHF calculations (all the orbitals are doubly occupied!). There are two wfn manipulations:
- ROHF: change occupation number of singly occupied orbitals
- MO 5 MO 0.0 OCC NO = 2.0000000 ORB. ENERGY = 0.0
- MO 5 MO 0.0 OCC NO = 1.0000000 ORB. ENERGY = 0.0
- ROHF and CASSCF: specify orbital contribution to MS (in fact twice). The 0.0 after MO should be changed to -1 (β), 0 (mixing true singlet), 1 (α), example
- MO 5 MO 1.0 OCC NO = 1.0000000 ORB. ENERGY = 0.0
sym_wfn
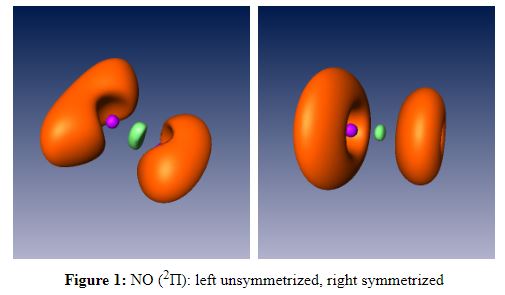
Molecular ab initio programs use basis real functions, in the case of linear molecules these computed MOs are not eigenfunctions of the Lz operator and therefore the cylindrical symmetry can be lost. This is particularly the case for open shell systems in a Π state. sym_wfn as been wriiten to correct this drawback for 2Π states calculated with ROHF. In order to get symmetrized figures run sym_wfn before performing the grid09 calculation. The program prompts the user to get the input wfn file name and the name of the symmetrized sym_wfn file.
mod_wfn
For large molecular systems mod_wfn enables to restrict the analysis to a subpart of the system. The atoms of the subpart of interest and their first neighbours are entered as data of mod_wfn
mod_wfn input
- filein (a40) : wfn file name
- natoms2 (free format) : number of selected atoms
- iatom(i),i=1,natoms2 : list of the selected atoms
- fileout (a40) : modified wfn file name
Example: CH2ClCH2COOH
The selected subpart is the functional group. The atoms in the wfn appear in the following order:
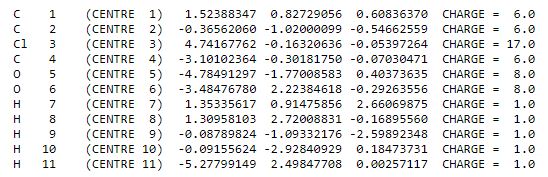
The selected atoms are C(2), C(4), O(5), O(6) and H(11). The input is :
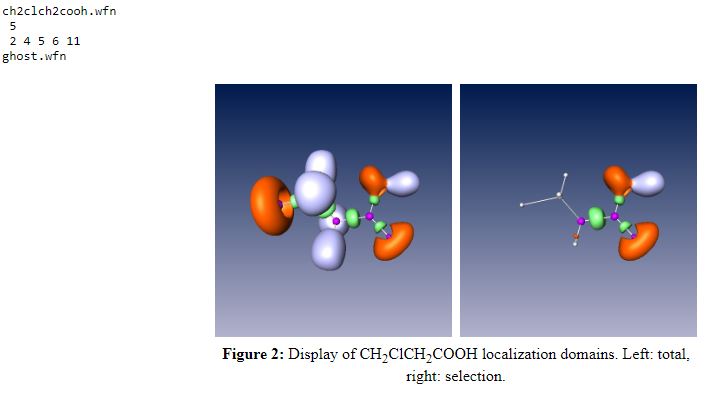
basin_prob
- filesij (a40) : esij.sbf file name
- ngroup : number of basins to be merged (if 0 stop)
- nelec(i),i=1,ngroup : label of the basins to be merged