Il y a deux possibilités de choix d'orbitales de départ : les deux
électrons sont soit sur la liaison à l'intérieur de la maille
élémentaire, soit sur l'un des atomes de la maille
(alternance entre des ions H![]() et H
et H![]() ). Dans les deux cas, les
itérations par l'IC des monoexcités produisent une fonction d'onde
Hartree-Fock d'une symétrie réduite, qui garde le caractère des
orbitales initiales (publication [11]). Pour construire une
solution avec la symétrie correcte il faut superposer une solution avec son
équivalent centré sur l'atome voisin. Les trois solutions différentes
correspondent à des structures différentes de la matrice de la densité,
dont je montre un morceau comprenant deux mailles à deux atomes :
). Dans les deux cas, les
itérations par l'IC des monoexcités produisent une fonction d'onde
Hartree-Fock d'une symétrie réduite, qui garde le caractère des
orbitales initiales (publication [11]). Pour construire une
solution avec la symétrie correcte il faut superposer une solution avec son
équivalent centré sur l'atome voisin. Les trois solutions différentes
correspondent à des structures différentes de la matrice de la densité,
dont je montre un morceau comprenant deux mailles à deux atomes :
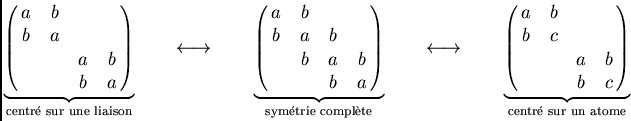 |
(6) |
true cm

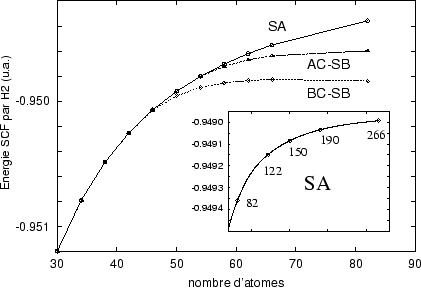 |
Les deux solutions Hartree-Fock à symétrie brisée ont des énergies plus basses que la solution représentant la symétrie de la molécule. L'effet ne se manifeste qu'à partir d'une certaine taille; pour des petits anneaux les trois points de départ amènent au même résultat.[3]Les brisures de symétrie ainsi que des conditions de stabilité ont été formulées par Thouless[#!Thouless!#] et détaillé par Paldus et Cizek dans une série d'articles dans les années 1970.[#!CizekPaldus!#]
Nous voyons ainsi que l'approche choisie pour resoudre les équations Hartree-Fock nous laisse la liberté d'obtenir des solutions dont la nature peut être anticipée; nous ne sommes pas forcés dans l'une ou dans l'autre solution par la procédure.
La brisure de symétrie est un artefact de la méthode Hartree-Fock due à la non-linéarité des équations. Pour obtenir un état fondamental de la symétrie de la molécule, il faut considérer la corrélation électronique, c'est à dire aller au delà de l'approximation Hartree-Fock. Mais même la perturbation au quatrième ordre (MP4) ou la sommation infinie des diagrammes de perturbation des diexcités (LCCD ou CEPA-0) ne suffisent pas à retrouver une solution unique.[4]Nous n'avons pas encore besoin d'une approche spécifique à la corrélation dans les solides, un calcul en orbitales canoniques par un programme standard suffit.
true cm
 |
Les méthodes Monte Carlo peuvent donner une nouvelle ouverture vers ce genre
de problématiques, puisqu'à partir d'une fonction d'onde approximative la
variante Diffusion Monte Carlo (DMC) est en principe capable d'extraire la
fonction d'onde de l'état fondamental et son énergie totale avec grande
précision. La source principale d'erreurs systématiques est la position
des n![]() uds (endroits dans l'espace
uds (endroits dans l'espace
![]() , où la
fonction d'onde est égale à zéro) de la fonction d'onde de départ.
Avec les trois solutions Hartree-Fock différentes, nous aurions une
estimation de l'erreur liée à la contrainte d'utiliser des n
, où la
fonction d'onde est égale à zéro) de la fonction d'onde de départ.
Avec les trois solutions Hartree-Fock différentes, nous aurions une
estimation de l'erreur liée à la contrainte d'utiliser des n![]() uds
(fixés à priori) de la fonction d'onde de départ pour la simulation DMC.
uds
(fixés à priori) de la fonction d'onde de départ pour la simulation DMC.
Un autre exemple de brisures de symétrie, cette fois plus réaliste que celui observé dans les systèmes fictifs tels que des anneaux d'hydrogène, sont les modifications tridimensionnelles du phosphore, où dans une structure cubique les trois électrons de valence de chaque atome s'apparient pour former des solides déformés et isolants (phosphore violet de Hittorf ou phorphore noir), ou bien se délocalisent pour former la structure électronique du phosphore métallique dans une structure cubique.
Dans les publications [6], [7] et [11] la construction des orbitales localisées est décrite en détail, pour des anneaux d'hydrogène, métallique et moléculaire, des anneaux d'atomes de béryllium et des hydrocarbures cycliques simples. Des applications à des systèmes réellement étendus ont été présentées dans la publication [14].