Un calcul Hartree-Fock (ou sans interaction biélectronique explicite :
Extended Hückel) fournit généralement des orbitales moléculaires
complètement délocalisées, ![]() , en diagonalisant l'opérateur de
Fock F avec des valeurs propres
, en diagonalisant l'opérateur de
Fock F avec des valeurs propres ![]() ; ces orbitales sont
développées sur des orbitales atomiques
; ces orbitales sont
développées sur des orbitales atomiques ![]() locales
locales
| (1) |
La fonction d'onde
![]() étant construit par un déterminant en fonction des positions
des électrons
étant construit par un déterminant en fonction des positions
des électrons ![]() et de leurs spins
et de leurs spins ![]()
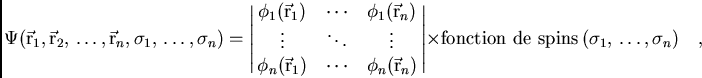 |
(2) |
les orbitales peuvent être localisées -- a posteriori -- par une transformation unitaire à l'aide d'une fonctionnelle ou par projection, sans changer cette fonction d'onde. La recherche des méthodes de localisation soit pour interpreter le sens chimique des orbitales soit pour rendre les calculs de corrélation moins long amenait au développement de plusieurs voies différentes dont je présenterai une sélection -- un article de revue plus complet est par exemple la réference RamirezSolis.
S. F. Boys décrit en 1960 une méthode de localisation,[#!Boys!#] qui
essaie de maximiser la distance moyenne entre les barycentres des orbitales.
Dans le même article Boys démontre déjà que la solution obtenue n'est
pas toujours unique : il existe par exemple un ensemble continu de solutions
pour les orbitales de valence d'une molécule de benzène. Dans une
molécule de monoxyde de carbone, trois orbitales de forme ``bananoïde''
représentent la triple liaison ![]() C
C![]() O
O![]() . Par contre, les paires
libres d'électrons se localisent comme le prévoit le modèle VSEPR de
Gillespie[#!Gillespie!#] et al., et l'on retrouve des concepts généraux
de la chimie. Il y a un petit prix à payer pour localiser : pour
déterminer les barycentres des orbitales il faut disposer des intégrales
du moment dipolaire
. Par contre, les paires
libres d'électrons se localisent comme le prévoit le modèle VSEPR de
Gillespie[#!Gillespie!#] et al., et l'on retrouve des concepts généraux
de la chimie. Il y a un petit prix à payer pour localiser : pour
déterminer les barycentres des orbitales il faut disposer des intégrales
du moment dipolaire
![]() , en plus des
intégrales utilisés pour le calcul Hartree-Fock.
, en plus des
intégrales utilisés pour le calcul Hartree-Fock.
Une autre méthode, dont nous nous servirons également par la suite, a
été proposée par J. Pipek et P. Mezey.[#!PipekMezey!#] Elle vise à
minimiser la fragmentation des orbitales sur les différents centres
atomiques. Puisque les fragmentations sont l'inverse des populations
![]() cette localisation
n'utilise que la matrice de densité
cette localisation
n'utilise que la matrice de densité
![]() et les intégrales de recouvrement
et les intégrales de recouvrement
![]() . Les deux matrices,
. Les deux matrices, ![]() et
et ![]() , sont d'habitude disponibles et utilisées dans le calcul de la
fonction d'onde, ce qui fait qu'il n'y a pas d'intégrales supplémentaires
à évaluer. La localisation Pipek-Mezey respecte bien les séparations en
orbitales
, sont d'habitude disponibles et utilisées dans le calcul de la
fonction d'onde, ce qui fait qu'il n'y a pas d'intégrales supplémentaires
à évaluer. La localisation Pipek-Mezey respecte bien les séparations en
orbitales ![]() et
et ![]() des liaisons triples, mais elle ne fournit plus
des paires libres séparées. En effet dans le cas d'une molécule
possédant plusieurs paires libres comme H
des liaisons triples, mais elle ne fournit plus
des paires libres séparées. En effet dans le cas d'une molécule
possédant plusieurs paires libres comme H![]() O il n'y a aucune raison que
les paires libres occupent des endroits différents autour du même centre
atomique (modulo des éléments de symétrie).
O il n'y a aucune raison que
les paires libres occupent des endroits différents autour du même centre
atomique (modulo des éléments de symétrie).
 |
Les deux méthodes, en partant d'un ensemble d'orbitales canoniques et orthonormées, produisent par des rotations deux à deux des orbitales localisées, également orthonormées.[2]Une généralisation de la méthode de Boys est élaborée par S. Liu et al.[#!Liu!#] en levant cette contrainte d'orthonormalité.
Une alternative aux méthodes a posteriori comme celle de Boys ou Pipek-Mezey est d'inclure la localisation dès la première itération de la solution des équations Hartree-Fock. Ceci est possible par la méthode ``Super-CI'',[#!MCSCF!#] mise au point à Toulouse (Réf. Daudey et publication [7],[3]Les réferences entre crochets [ ] sont détaillées dans la partie V : Publications et ne figurent pas dans la liste des références à la fin de l'exposé des travaux. et indépendamment à Tokyo par T. Sano et O. Matsuoka[#!Sano!#]), applicable non seulement à des systèmes moléculaires, mais également aux systèmes périodiques, à condition d'introduire quelques approximations.
Avant de décrire l'approche ``Super-CI'', nous devons mentionner l'inclusion d'un potentiel de localisation par projection dans la matrice Fock d'un système périodique, proposée par A. Shukla et al.[#!Shukla!#] L'idée est d'ajouter à l'opérateur de Fock de la maille élémentaire des projecteurs sous forme de multiplicateurs de Lagrange, qui introduisent la condition d'orthogonalité des orbitales de la maille élémentaire aux orbitales des mailles voisines. En diagonalisant cet opérateur de Fock modifié, on obtient des orbitales ``canoniques'' de la maille élémentaire, seulement délocalisées à l'intérieur de cette maille. La périodicité est prise en compte, à la fois dans les projecteurs et dans les sommations infinies des intégrales de Coulomb et d'échange lors de la construction de l'opérateur de Fock. Les applications publiées concernent le trans-polyacétylène (1D) et LiF et LiH (3D).[#!ShuklaII!#]
Revenons à la méthode ``Super-CI'', qui introduit une voie alternative de construction des orbitales localisées sans passage par les fonctions de Bloch : l'idée centrale est de baisser l'énergie totale d'un déterminant donné par une interaction de configurations dans l'espace des configurations mono-excitées et de construire un schéma d'itération par deux approximations :
La deuxième approximation permet de rendre le calcul aussi efficace qu'une
diagonalisation directe de l'opérateur de Fock, mais avec la possibilité
d'obtenir directement des orbitales localisées à partir d'un jeu
d'orbitales localisées approchées. Si par exemple les orbitales de
départ sont issues d'un calcul en base minimale ou construites ``à priori'', la
solution obtenue à la fin des itérations conserve le plus possible leur
caractère initial. Un avantage supplémentaire tient à ce que la
procédure peut être facilement appliquée aux systèmes étendus, en
cherchant des orbitales moléculaires centrées dans une maille
élémentaire, avec des queues décroissantes sur les mailles voisines.
Dans les molécules, l'orthogonalité entre orbitales occupées et
orbitales virtuelles est assurée dès le départ, parce que les
corrections
![]() pour les
occupées et
pour les
occupées et
![]() pour les
virtuelles laissent les deux espaces bien séparés. Par contre, dans un
système étendu avec les orbitales localisées dans des mailles
pour les
virtuelles laissent les deux espaces bien séparés. Par contre, dans un
système étendu avec les orbitales localisées dans des mailles ![]() , soit
l'orthogonalité soit la symétrie translationnelle sont détruites par la
génération simultanée des nouveaux coefficients des orbitales occupées
et virtuelles :
, soit
l'orthogonalité soit la symétrie translationnelle sont détruites par la
génération simultanée des nouveaux coefficients des orbitales occupées
et virtuelles :
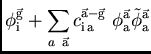 |
(4) |
Pour conserver la symétrie translationnelle il faut considérer
uniquement les coefficients des fonctions de base de la maille
d'origine
![]() et orthogonaliser les orbitales à chaque itération.
L'orthogonalisation est effectuée par un processus iteratif, utilisant le
développement en série de la matrice
et orthogonaliser les orbitales à chaque itération.
L'orthogonalisation est effectuée par un processus iteratif, utilisant le
développement en série de la matrice
![]() . Le calcul direct par inversion est impossible parce que
S est d'ordre infini.
. Le calcul direct par inversion est impossible parce que
S est d'ordre infini.
Avant d'aborder le problème de la corrélation dans les systèmes périodiques tridimensionnels infinis, il me semblait plus raisonnable de développer les méthodes de corrélation dans des molécules périodiques unidimensionnelles et limitées dans l'espace. Les conditions aux limites cycliques (ou de Born-von Karmann) utilisées dans tout les calculs périodiques infinis doivent être également satisfaites par des chaînes fermées en anneaux, même de petite taille. Un atome aussi peut être vu comme un anneau, moins trivialement un dimère. Une fois les méthodes établies pour ces applications, la généralisation aux systèmes infinis semble directe, puisqu'on peut toujours énombrer les mailles d'un réseau infini, partant d'une maille de référence. Le passage d'un anneau à un système tridimensionnel infini se fait en changeant le nombre et les indices des mailles voisines de la maille de référence.
true cm
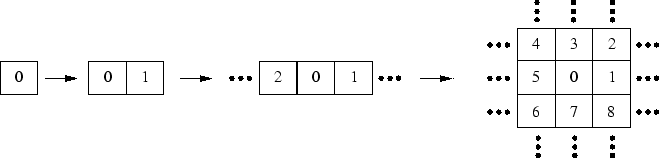 |
Le jeu d'orbitales centré sur la maille de référence détermine
alors toutes les orbitales moléculaires du système périodique, les
coefficients de développement sur les orbitales atomiques portant de plus un
indice de maille ![]() :
:
| (5) |
En introduisant des rayons de coupure dans les indices ![]() et dans les
éléments de la matrice Fock, ainsi que dans la matrice
d'IC dont on veut déterminer la valeur propre la plus basse, les
itérations sont effectuées avec les étapes suivantes :
et dans les
éléments de la matrice Fock, ainsi que dans la matrice
d'IC dont on veut déterminer la valeur propre la plus basse, les
itérations sont effectuées avec les étapes suivantes :