This input module describes the overall type of calculation that is to
be done. It also contains four submodules describing the
performance of parallel calculations, the control of the two different geometry optimization
routines, as well as control the general routines for calculating
numerical geometrical derivatives of molecular energies or selected
first- and second-order properties. We note that this input module has
to start all input files for DALTON.
-
.DIRECT
- The calculation is to be done in a direct
manner, that
is, the two-electron integrals are to be
constructed ``on the fly''
and not written to disc as is the default. This keyword will only work
for SCF wave functions,
Density Functional Theory
calculation and for
coupled cluster (CC) calculations. In
HF and DFT calculations the
two-electron integrals (and differentiated
two-electron integrals) will not be written to disc in any part of the
calculation, whereas in the direct CC approach, the two-electron
integrals will be stored in three general-indexed batches, thus
requiring a loop over all basis functions for one of the two-electron
integral indices [].
-
.DOUGLAS-KROLL
- Include scalar relativistic effects by using the
Douglas-Kroll transformed one-electron potential and kinetic energy Hamiltonian.
-
.INPTES
- Test the input of the
**DALTON INPUT input
module. The program will abort after the completion of the input test,
and no calculation will be executed.
-
.INTEGRALS
- Invoke the HERMIT and/or the ERI program for generating molecular one-
and two-electron integrals. See
Chapter 22 for the HERMIT and Section 22.2.8 ERI programs, respectively.
-
.ITERATION
READ (LUCMD, '(I5)') ITERNR
Tells the program at which iteration to start the geometry optimization
using the
**WALK module.
Note that this will not affect which molecule
input file that is going to be read, as this is handled by the
job script dalton. It only determines what number the
output of the predicted molecular geometry will be, as well as where to start
writing information on the files containing information about an IRC
calculation (DALTON.IRC) or a dynamical trajectory walk (DALTON.TRJ).
-
.MAX IT
READ (LUCMD, '(I5)') ITERMX
Change the maximum number of geometry iterations
that can be done. Default
is 20. For numerical
differentiation/vibrational averaging, the number of iterations will
be reset to 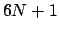 (where
(where 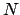 is the number of nuclei) as the number
of required iterations for these calculations are well
defined. This number has to be increased in Intrinsic Reaction Coordinate
(IRC) or dynamical
trajectory studies. However, changing this variable will override this reset
option.
is the number of nuclei) as the number
of required iterations for these calculations are well
defined. This number has to be increased in Intrinsic Reaction Coordinate
(IRC) or dynamical
trajectory studies. However, changing this variable will override this reset
option.
-
.OPTIMIZE
- Do a geometry walk. If no
input is given in the
*OPTIMIZE input submodule, an optimization of the molecular
geometry to a stationary point with no
negative Hessian eigenvalues (a
local minimum) will be done using the default first-order
methods. However, this may be changed using appropriate
keywords in the submodule
*OPTIMIZE, and we refer to examples in
the chapter on potential energy surfaces
(Chapter 7), and subsection 21.1.3
describing the input cards for the
*OPTIMIZE submodule for a more
detailed description of possible options.
-
.NMDDRV
- Calls for a generalized numerical geometry
differentiation. These routines will take advantage of the full
molecular point group in order to minimize the number of point to be
calculated. What order of derivatives and whether any analytical
derivatives are to be used is determined in the
*NMDDRV input
module.
-
.PARALLEL
- Requests that the calculation of two-electron
integrals
is to be done in parallel. This also
implies that the calculation is
done without writing two-electron integrals to disc. This keyword only
applies to SCF wave
functions and DFT calculations, but all two-electron integral
evaluations in an SCF calculation will be done parallel as well as the integration of the exchange-correlation functionals and kernels. More details
about the parallelization strategy in DALTON can be found
in Ref. [90].
The keyword requires that the program has been installed and compiled
with the appropriate preprocessor directives for an MPI
installation.
Note that
in order to evaluate the parallelization efficiency, a print level of
at least 2 is needed in the
*PARALLEL submodule.
-
.PARNMD
- Option non-functional. Do not use.
-
.PRESORT
- Requests that the
two-electron integrals should be
sorted and that the integral transformation routines of Bjørn
Roos should be used during execution of the program. The keyword is needed if one attempts an MCSCF, CI or MP2 (run through the SIRIUS module and not using the Coupled-Cluster module) with more than 255 basis functions.
-
.PRIERR
READ (LUCMD, *) IPRERR
Reads in the print level that is to be used in the DALTON.ERR
file. Default print level is IPRUSR+1.
-
.PRINT
READ (LUCMD, *) IPRUSR
Reads in the print level that is to be used the rest of the subsequent
calculations. Default is a print level of 0.
-
.PROPERTIES
- Invoke the ABACUS program for the evaluation of static
and dynamic properties. See Chapter 25.
-
.RESPONSE
Invoke the RESPONSE program for the evaluation of static and dynamic
properties. See Chapter 26.
-
.RUN ALL
Invoke all the programs HERMIT, SIRIUS, RESPONSE, and ABACUS for a single point
calculation.
-
.RUN PROPERTIES
Invoke the programs HERMIT, SIRIUS, and ABACUS for a single point
calculation.
-
.RUN RESPONSE
Invoke all the programs HERMIT, SIRIUS, and RESPONSE for a single point
calculation.
-
.RUN WAVE FUNCTIONS
Invoke the programs HERMIT and SIRIUS for a single point energy calculation.
-
.RUNERI
- Force the use of the vectorized integral code ERI where possible.
-
.TOTSYM
- Consider only totally symmetric
perturbations.
This option only affects geometric perturbations calculated using the
second-order based
.WALK option and static
electric-field perturbations requested through the keyword
.POLARI.
-
.VECLEN
READ(LUCMD,*) IVECLN
Set the number of Fock matrices to be used during Fock-matrix constructions in
direct calculations. This function is only of interest for vector machines.
The default is 128. The larger the number, the more memory will be required
in the calculation.
-
.WALK
- Do a geometry walk. If no
input is given in the
*WALK input submodule, an optimization of the molecular
geometry to a stationary point with no
negative Hessian eigenvalues (a
local minimum) will be done using a second-order method with
analytical Hessians. However, this may be changed by appropriate
keywords in the submodule
*WALK, and we refer to examples in
the chapter on potential energy surfaces
(Chapter 7), and subsection 21.1.3
describing the input cards for the
*WALK submodule for a more
detailed description of possible options.
-
.WAVE FUNCTIONS
-
Invoke the SIRIUS program for the evaluation of SCF, MP2, Coupled
Cluster and MCSCF wave functions as well as DFT calculations. See
Chapter 24.