En utilisant des orbitales Kohn-Sham de la DFT au lieu d'orbitales Hartree-Fock, on obtient une décomposition analogue, mais tenant compte de la corrélation électronique intra-moléculaire et inter-moléculaire. Les étapes du calcul suivent le schéma proposé par M. Bickelhaupt et E. J. Baerends,[#!Bickelhaupt!#] et utilisé par ces auteurs pour décrire les liaisons chimiques et la liaison hydrogène.[#!Fonseca!#] Cette décomposition est implémentée dans le code ADF (Amsterdam Density Functional),[#!ADF!#] pour les fonctionnelles LDA,[#!LDA!#] BLYP[#!BLYP!#] et PW91.[#!PW91!#] Dans ce logiciel les orbitales moléculaires sont développées sur des bases numériques ou des bases de fonctions Slater.
Pour exploiter d'autres fonctionnelles, et pour pouvoir comparer les résultats avec les calculs SAPT, où la base de développement des orbitales moléculaires sont des fonctions gaussiennes, j'ai intégré le schéma dans le module SCF de mes programmes, en faisant appel aux routines DFT du package MOLPRO.[#!MOLPRO!#]
La comparaison avec les termes SAPT est directe pour la première étape,
qui fait interagir les densités électroniques des monomères sans
corrélation intramoléculaire (niveau Hartree-Fock) ou avec corrélation
intramoléculaire (niveau DFT). La décomposition en échange, dispersion
et induction est plus difficile parce que la dispersion n'a pas de place
explicite dans la DFT. Nous pourrons comparer la ``répulsion de Pauli'' avec
l'échange au premier ordre en SAPT (E![]() ) et l'interaction
orbitalaire avec la somme de dispersion et induction. Comme il n'y a pas
de fondement théorique rigoureux et commun entre les deux approches SAPT et
DFT, nous pouvons essayer des expériences numériques en regroupant des
termes différament.
) et l'interaction
orbitalaire avec la somme de dispersion et induction. Comme il n'y a pas
de fondement théorique rigoureux et commun entre les deux approches SAPT et
DFT, nous pouvons essayer des expériences numériques en regroupant des
termes différament.
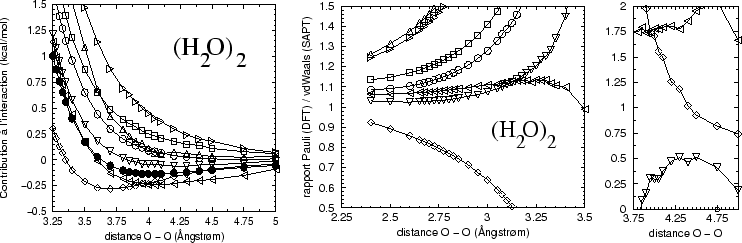
Nous avons observé ainsi un chose surprenante : parmis la multitude de fonctionnelles sur le marché, la fonctionnelle Perdew-Wang 91[#!PW91!#] semble reproduire la dispersion de SAPT dans le terme ``répulsion de Pauli''.
Il faut d'abord remarquer que ce terme n'est pas toujours positif pour cette fonctionnelle (Fig. 17), mais montre un petit creux comme un minimum van der Waals.
Alors nous pouvons comparer la ``répulsion de Pauli'' avec une somme de
termes en SAPT correspondant à des interactions van der Waals seulement,
sans interaction électrostatique ni induction
| (27) |
Le rapport entre
le terme ``répulsion de Pauli'' de la décomposition DFT et
la contribution ``van der Waals'' en SAPT est rélativement proche
de 1 pour la fonctionnelle PW91, contrairement aux autres fonctionnelles, du
moins lorsque la distance intermoléculaire n'est pas trop
longue. Ce rapport n'est plus une bonne quantité lors du passage
par zéro de ![]() de SAPT, et au delà la précision du calcul
d'un coté, et l'incapacité de la DFT de décrire des éffets de
dispersion à longue portée de l'autre coté font que la comparaison a des
limites.
true cm
de SAPT, et au delà la précision du calcul
d'un coté, et l'incapacité de la DFT de décrire des éffets de
dispersion à longue portée de l'autre coté font que la comparaison a des
limites.
true cm
 |
true cm
 |
La prise en compte des termes d'échange des corrections de corrélation
intramoléculaire à la dispersion (
![]() et
et
![]() ),
indisponible dans le programme actuel, pourrait corriger la
contribution van der Waals et améliorer le rapport ``répulsion de Pauli
(DFT) / van der Waals (SAPT)''.
),
indisponible dans le programme actuel, pourrait corriger la
contribution van der Waals et améliorer le rapport ``répulsion de Pauli
(DFT) / van der Waals (SAPT)''.
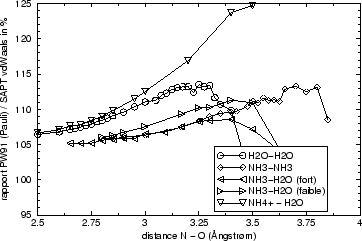
Nous avons observé le même effet pour cinq dimères différents et nous doutons que le résultat soit dû à une pure coincidence, même si nous sommes pour l'instant incapables de donner une raison plus fondée.
En fait nous sommes conscients que la méthode SAPT telle qu'elle existe actuellement ne contient pas tous les termes de corrélation intramoléculaires. Il nous a donc paru intéressant d'avoir une idée de la valeur des termes manquants dans cette méthode. Nous pouvons même imaginer une méthode ``hybride'' qui contiendrait des termes SAPT et des termes qui seraient calculés comme différences DFT/SAPT, voir le tableau VII.
Afin de vérifier nos hypothèse nous avons appliqué les expressions du
tableau VII au dimère de NH![]() du chapitre précédent,
pour lequel certains termes SAPT ont été recalculé. Parmi les nombreuses
fonctionnelles disponibles nous avons retenu dans cette étude BLYP,
PW91 et PBE0. Cette dernière est construite à l'aide de conditions
analytiques pour faibles et fortes densités et elle inclue un terme d'échange
exact.[#!PBE!#] En absence de critères de sélection plus fondés,
nous pourrons expérimenter et voir, si la reconstruction des contributions
de SAPT est faisable en combinant les résultats des différentes
fonctionnelles.
du chapitre précédent,
pour lequel certains termes SAPT ont été recalculé. Parmi les nombreuses
fonctionnelles disponibles nous avons retenu dans cette étude BLYP,
PW91 et PBE0. Cette dernière est construite à l'aide de conditions
analytiques pour faibles et fortes densités et elle inclue un terme d'échange
exact.[#!PBE!#] En absence de critères de sélection plus fondés,
nous pourrons expérimenter et voir, si la reconstruction des contributions
de SAPT est faisable en combinant les résultats des différentes
fonctionnelles.
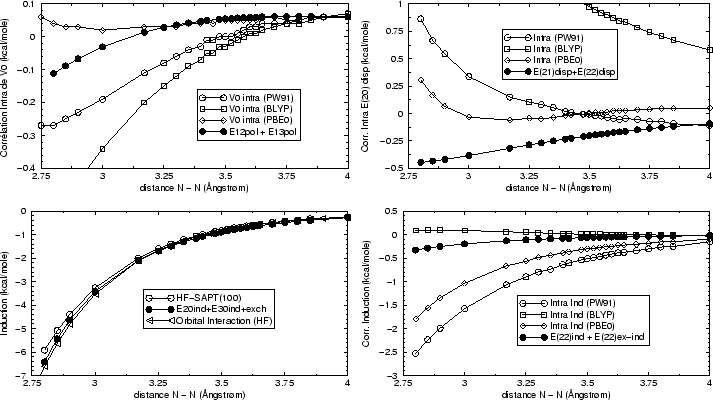
Les approximations apportées sont (Figure 19) visiblement
trop importantes pour justifier une modélisation directe des contributions SAPT
coûteuses. D'autre part, la figure montre qu'une fois de plus le choix de
la fonctionnelle a une grande influence sur les formes des contributions. Une
analyse plus fine permettra peut-être un jour d'estimer l'ordre de grandeur
et le signe des contributions SAPT manquantes (d'échange ou d'ordres
supérieurs), ou bien, de façon complémentaire de construire de
nouvelles fonctionnelles en profitant des
données présentées ici. Toutefois nous pouvons constater que l'ordre de
grandeur de l'erreur sur les différents termes est d'environ
0.25![]() kcal/mole pour les deux premières reconstructions de la figure
19, et plus grand pour la corrélation intramolculaire
sur le terme d'induction.
kcal/mole pour les deux premières reconstructions de la figure
19, et plus grand pour la corrélation intramolculaire
sur le terme d'induction.
 |
Finalement nous pourrons construire ainsi l'interaction complète à partir
des reconstructions de la figure précédente, en utilisant les résultats
les plus proches de la référence SAPT. Cela revient à choisir la
corrélation intramoléculaire de V![]() par V
par V![]() (intra) PW91, la
corrélation intramoléculaire de la dispersion PBE0 l'interaction
orbitalaire de Hartree-Fock, et finalement l'interaction orbitalaire BLYP
pour introduire la corrélation intramoléculaire de l'induction. A cela
s'ajoutent les contributions à calculer par Hartree-Fock et SAPT du tableau
VII.
(intra) PW91, la
corrélation intramoléculaire de la dispersion PBE0 l'interaction
orbitalaire de Hartree-Fock, et finalement l'interaction orbitalaire BLYP
pour introduire la corrélation intramoléculaire de l'induction. A cela
s'ajoutent les contributions à calculer par Hartree-Fock et SAPT du tableau
VII.
Cette recette élaborée à l'aide de (NH![]() )
)![]() devrait être
confirmée par des calculs sur d'autres systèmes. Mais la variation
des résultats en fonction du choix de la fonctionnelle est tellement grande,
qu'il faudrait peut-être attendre l'élaboration d'autres fonctionnelles
specifiquement conçues pour des interactions intermoléculaires comme le
PBE0AC[#!PBE0AC!#] avec corrections asymptotiques.
devrait être
confirmée par des calculs sur d'autres systèmes. Mais la variation
des résultats en fonction du choix de la fonctionnelle est tellement grande,
qu'il faudrait peut-être attendre l'élaboration d'autres fonctionnelles
specifiquement conçues pour des interactions intermoléculaires comme le
PBE0AC[#!PBE0AC!#] avec corrections asymptotiques.
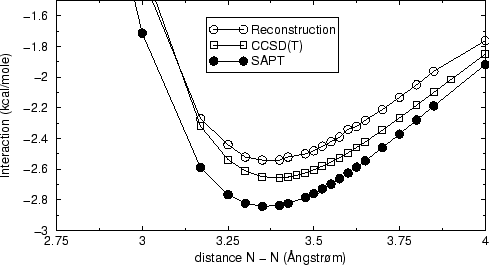
La figure 20 montre que le mélange de fonctionnelles et SAPT
peut conduire à une courbe de potentiel raisonnable, malgré les
différences entre SAPT et ses reconstructions pour les contributions
individuelles de figure 19. Les différences d'énergie,
par rapport à CCSD(T) supermoléculaire et SAPT sont de l'ordre de
0.5![]() kcal/mole et la position du minimum est au même endroit avec les
trois méthodes.
kcal/mole et la position du minimum est au même endroit avec les
trois méthodes.
 |
Cette voie de recherche empirique peut néanmoins apporter du sens physique aux démarches théoriques rigoureuses, présentées dans le chapitre précédent. L'approche CCSD(T) que nous avons utilisée comme référence du potentiel intermoléculaire ne fournit que ce potentiel total, sans aucune information supplémentaire.
La DFT étant une méthode parametrée en cherchant de respecter des valeurs limites et de reproduire le plus possible la densité et l'énergie de l'état fondamental d'un système ne donne pas non plus à priori des détails des potentiels intermoléculaires.
Ce n'est qu'en interaction avec d'autres méthodes tels que SAPT ou les méthodes Monte Carlo et modèles physiques (n'oublions pas l'expérience) que les fonctionnelles utilisées quotidiennement peuvent être améliorées et que leur capacités peuvent être discutées.
=1
