Présentation de l'interface
Zoom
Pour optimiser l’affichage de l’interface pensez à utiliser le zoom du navigateur (touche Ctrl et mollette de la souris ou Ctrl et les touches + ou -).
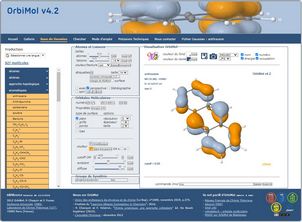
Visualisation OrbiMol
La rubrique « Visualisation OrbiMol » permet de changer la couleur du fond de la fenêtre Jmol, les données affichées lorsque qu’on active l’affichage d’une orbitale moléculaire : le nom de la molécule, l’énergie, le numéro et l’occupation de l’OM.
Dans le cas des radicaux, la HOMO est en fait semi-occupée (SOMO). Dans les molécules à l’état triplet (ex. O2) la HOMO et la HOMO-1 sont semi-occupées. En cas de doute, la charge et la multiplicité peuvent être lues dans le fichier de résultats (rubrique " Fichier Gaussian ")
L’énergie est calculée en unité atomique (u.a. ou Hartree). Les facteurs de conversion sont :
(valeurs utilisées dans Gaussian 03)
L’occupation indique si l’orbitale affichée est occupée « O » (par 1 ou 2 électrons) ou vacante « V ».
Le choix de la résolution de la fenêtre de visualisation permet d’intervenir indirectement sur la taille de la capture d’écran.
Source wikipedia
 |
Une sortie VRML est disponible pour réaliser une impression 3D d'une orbitale moléculaire. La procédure est expliquée ici. |
Un clic droit de la souris sur la fenêtre Jmol permet d’afficher le menu " standard " Jmol. La ligne de commande permet un accès rapide aux commandes de Jmol.
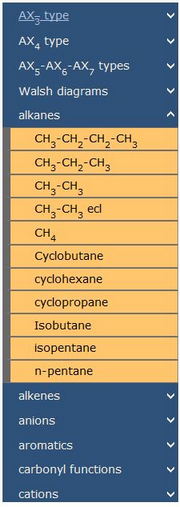
Menu d’OrbiMol et recherche dans la base de données
|
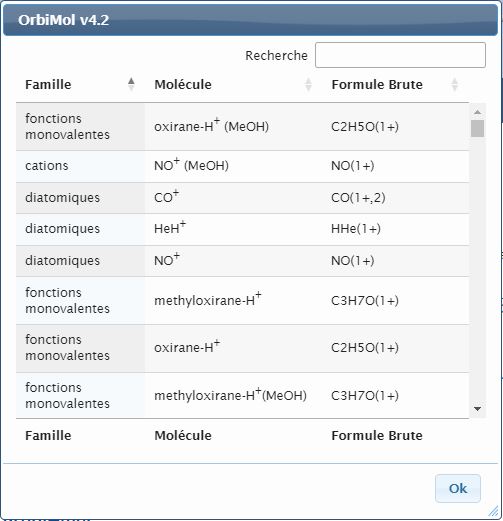
Menu d’OrbiMol : les molécules sont organisées par fonctions organiques, type VSEPR, ... une molécule peut faire partie de plusieurs rubriques. Ces rubriques pourront évoluer au gré des ajouts demandés par les utilisateurs de cette base de données. Depuis la version 3.1, il est possible d'effectuer une recherche dans la base de données. Vous pouvez lancer une recherche en cliquant sur " Chercher " de la barre de navigation ou sur le nombre de molécules du menu d'OrbiMol. La recherche s'effectue sur les familles, les noms ou les formules brutes des molécules. |
|
Fichier Gaussian/fréquences/population
Quand une molécule est lue par l'interface d'OrbiMol, un lien " Fichier gaussian : " suivit du nom de la molécule apparaît dans la barre de navigation. Cela permet de lire le fichier résultat ainsi que de le sauvegarder pour le lire avec d’autres programmes (clic droit de la souris et option " enregistrer la cible sous … "). Les calculs Gaussian/fréquences sont réalisés avec le programme de chimie théorique commercial, Gaussian version 03. Les calculs ont été généralement effectués au niveau semi-empirique AM1. La géométrie a été optimisée au même niveau, sauf pour de petites molécules pour lesquelles la géométrie expérimentale a été utilisée.
Les calculs de la fonction ELF ont été réalisés par la suite logiciel TopMod développée dans notre laboratoire. Ces calculs ont été réalisés en B3LYP avec une base d'orbitales 6-31+G(d).
Atomes et liaisons
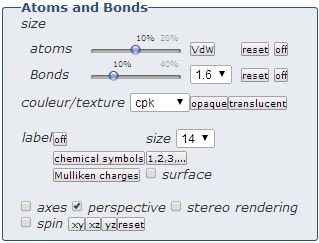
La rubrique " Atomes et Liaisons" permet de modifier les options du modèle moléculaire. Les options sont les suivantes pour la représentation boules/bâtons :
|
|
" taille des atomes" est un curseur permettant de modifier proportionnellement le rayon atomique des atomes (par défaut 10% du rayon de Van der Waals) accompagné du bouton " VdW " pour afficher 100% du rayon de Vand der Waals, " reset " pour revenir à la valeur par défaut et d'un bouton " off " pour le mettre à zéro, |
|
|
" taille des liaisons " même chose que l’option précédente pour les liaisons, |
|
|
" couleur/texture " permet de choisir pour la couleur du modèle (cpk : couleur attribuée par élément chimique, rouge, bleu, vert, scian, b&w), accompagné de deux boutons pour afficher le modèle opaque (par défaut) ou transparent, |

|
|
" étiquettes " affiche les symboles des éléments (nomenclature de Berzelius), les symboles avec des numéros, ou les charges de Mulliken avec le choix de la taille de la fonte et une projection des charges de Mulliken sur la surface de Connolly (explications ici) avec le code couleur roygb de Jmol. |
|
|
Le calcul des valeurs de la charge de Mulliken sur les points de la surface se fait par la formule : 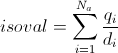 où Na est le nombre d'atomes, qi est la charge de Mulliken de l'atome i et di la distance entre l'atome i et le point de la surface. où Na est le nombre d'atomes, qi est la charge de Mulliken de l'atome i et di la distance entre l'atome i et le point de la surface.
|
|
|
|
" axes " affiche le repère cartésien xyz, |
|
|
" perspective " affiche avec perspective (par défaut), |
|
|
" 3D " permet de passer au mode stéréoscopique (sans lunettes), |
|
|
" spin " pour une rotation infinie, |
|
|
Placement perpendiculairement aux plans " xy " ou " xy " ou " yz " et un bouton " reset " pour revenir à la position par défaut. |
Orbitales Moléculaires
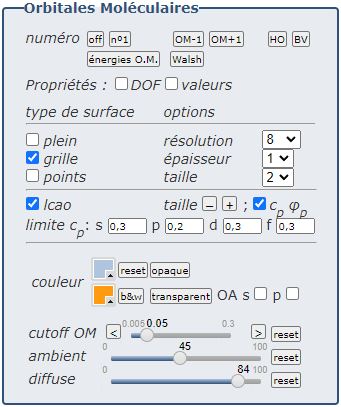
La rubrique " Orbitales Moléculaires " permet de choisir l’OM à visualiser ainsi que les options d’affichage. Par défaut la HOMO est affichée sauf si l'option off a été choisie :
|
|
" numéro " permet de choisir l’O.M. par ordre d’énergie, à savoir, " n°1 " la plus basse en énergie, la " précédente " (OM-1) et la " suivante " (OM+1) dans le diagramme énergétique ou par critère d’occupation (cf. rubrique " Visualisation OrbiMol ") comme la " HOMO " (highest occupied molecular orbital) et la " LUMO " (lowest unoccupied molecular orbital). | ||||||
|
|
" diagramme des énergies orbitalaires " permet d'afficher les informations sur la molécule (formule brute et développée), sur son état électronique (charge, multiplicité de spin) et sur le diagramme énergétiques des OM. Les niveaux sont cliquables pour afficher l'isosurface de l'OM correspondante.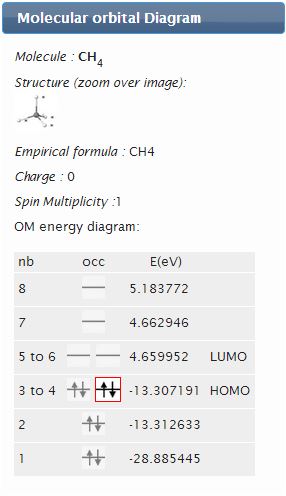
|
||||||
|
|
" Walsh " permet d'afficher les diagrammes de Walsh.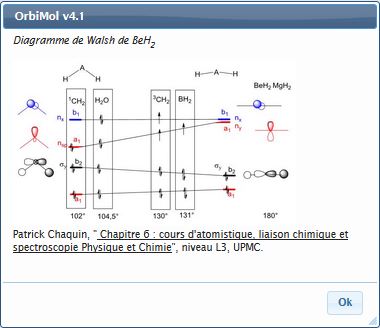
|
||||||
|
|
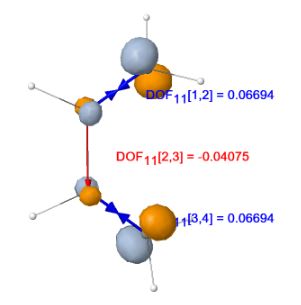
DOF " permet d'afficher les forces orbitalaires dynamiques.
La dérivée des énergies des orbitales moléculaires par rapport à une longueur de liaison (forces orbitalaires dynamiques [DOF]) est utilisée pour estimer le caractère de liant / antiliant des MO de valence le long de cette liaison.
|
||||||
|
|
" type de surface " permet de choisir la manière de représenter l'isosurface des OMs en mode grille, plein ou points. Ces trois types sont combinables. Les options sont disponibles pour modifier l'apparence des OMs mais pas des OAs (LCAO) : la résolution de l'isosurface dans les 3 modes, l'" épaisseur " des lignes et la " taille des points " des points. Attention au choix de la résolution, car cela influence directement la rapidité d’affichage de la surface | ||||||
|
|
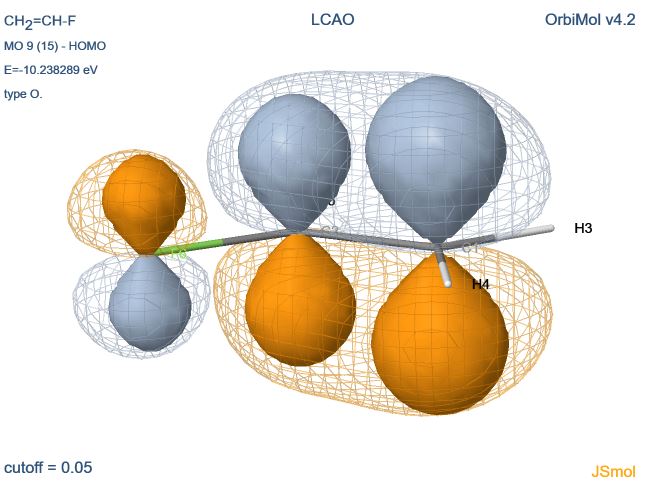
le type " LCAO " permet d'afficher les OAs utilisées dans le calcul LCAO (rubrique " Molecular Orbital Coefficients " dans la sortie Gaussian Inc.).
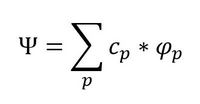
 |
||||||
|
|
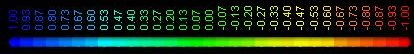
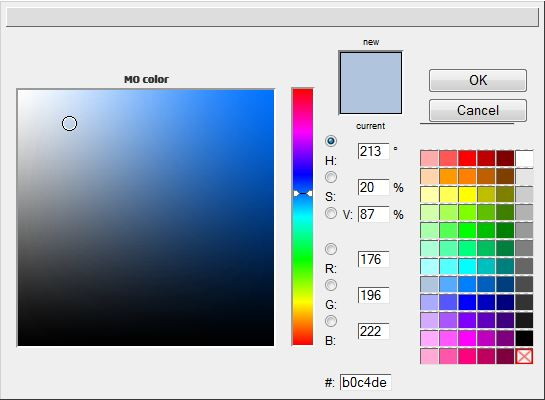
" couleur " modifie les couleurs des phases positives et négatives des OMs et des OAs (s, p),
accompagnées de boutons pour rendre les surfaces opaques (défaut) ou transparentes. L'option " b&w " passe toutes les surfaces (OMs, OAs) en noir et blanc. Un bouton " reset " permet de revenir aux couleurs "OrbiMol" par défaut.
La palette s'affiche de cette manière :
|
||||||
|
|
" cutoff OM " est un paramètre permettant d’affiche la valeur (absolue) de la fonction d’onde sur la surface d’isovaleur calculée. Une valeur élevée (0.2 ou plus) permet de voir la localisation principale de l’orbitale moléculaire. Une valeur plus faible (0.1-0.05) indique les localisations secondaires. | ||||||
|
|
" ambient " et " diffuse ", deux options, pour modifier la lumière ambiante et le rendu de la surface accompagnées de leurs boutons " reset " respectifs. |
Mode de vibrations
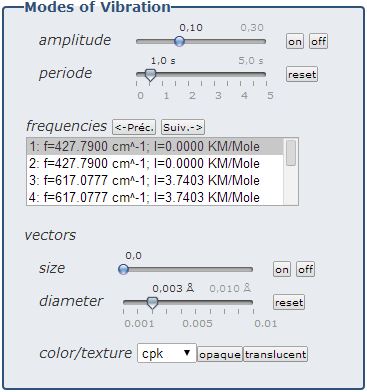
La rubrique " Modes de vibrations " permet d'animer les molécules en choisissant le mode de vibrations voulu.
|
|
" amplitue " permet de modifier l'amplitude de l'animation de la vibration. Le curseur est accompagné de deux boutons " on" et " off ", |
|
|
" période " permet de choisir le temps de la vibration (1 seconde par défaut) et un bouton " reset " pour revenir à la valeur par défaut, |
|
|
" fréquences et intensités " : le bouton " afficher la liste " montre la liste des fréquences pour une sélection ultérieure, les boutons " Préc." et " Suiv. " assure un parcours rapide de cette liste, |
|
|
" vecteurs " si l'animation n'est pas souhaitée, cette option permet de représenter les vibrations sur chacun des atomes. |
|
|
" taille " pour modifier la longueur des vecteurs, |
|
|
" diamètre " pour modifier le diamètre des vecteurs. |
Approche topologie de la liaison chimique
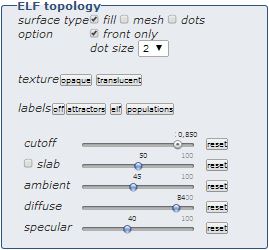
La rubrique " Approche topologique " permet d'afficher la fonction de localisation électronique, fonction ELF. Pour plus d'informations sur cette fonction c'est ici.
Le fichier contenant les populations est accessible par le lien qui apparaît en haut dans la barre de navigation.
|
|
" type de surface " permet de en mode grille, plein ou points. Ces trois types sont combinables. Les options sont disponibles pour modifier leur apparence : " face avant " pour les modes plein ou grille (pas pour le mode points) et " taille des points " pour le mode point, |
|
|
" texture " modifie la texture de l'isosurface, opaque (défaut) ou transparente. |
|
|
" étiquettes " affiche les noms des bassins C(Xi), V(Xi, Yi), V(Xi, Hi) (avec i les indices affichés dans le fichier contenant les populations), la valeur de la fonction ELF aux attracteurs et les valeurs des populations. |
|
|
" cutoff " est un paramètre permettant d’affiche l'isosurface de la valeur indiquée. Plus cette valeur est haute et plus la localisation électronique est élevée. |
|
|
" Slab " permet de couper l'isosurface. La valeur par défaut, 50, montre 50% de l'arrière de l'isosurface. |
|
|
" ambient ", " diffuse " et " specular ", trois options, pour modifier la lumière ambiante et le rendu de la surface accompagnées de leurs boutons " reset " respectif. |
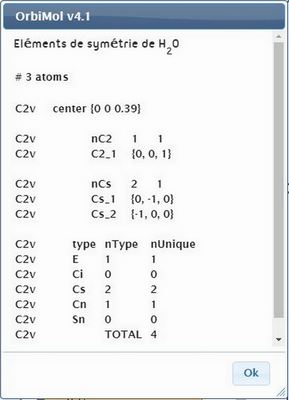
Groupe de symétrie
La rubrique " Groupe de symétrie " permet d'afficher les éléments de symétrie de la molécule.
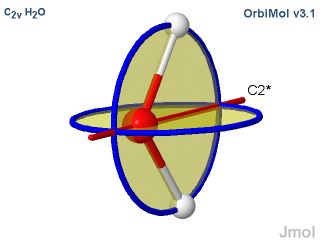
Enfin, vous pouvez afficher la liste des éléments de symétrie.