
|
|
Julia Contreras-García: Software |
| Press this badge for direct link to my Researcher ID |
Available here
Software to estimate the critical temperature of a superconductor through the determination of its networking value.
Developed by Trinidad Novoa, J. Contreras-García and Yvon Maday.
For more information see: https://doi.org/10.1038/s41467-021-25687-0
Available for download here
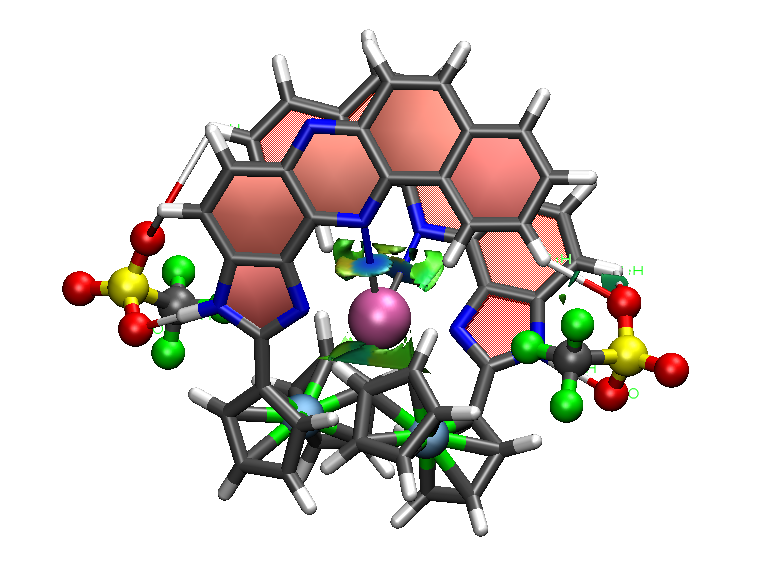
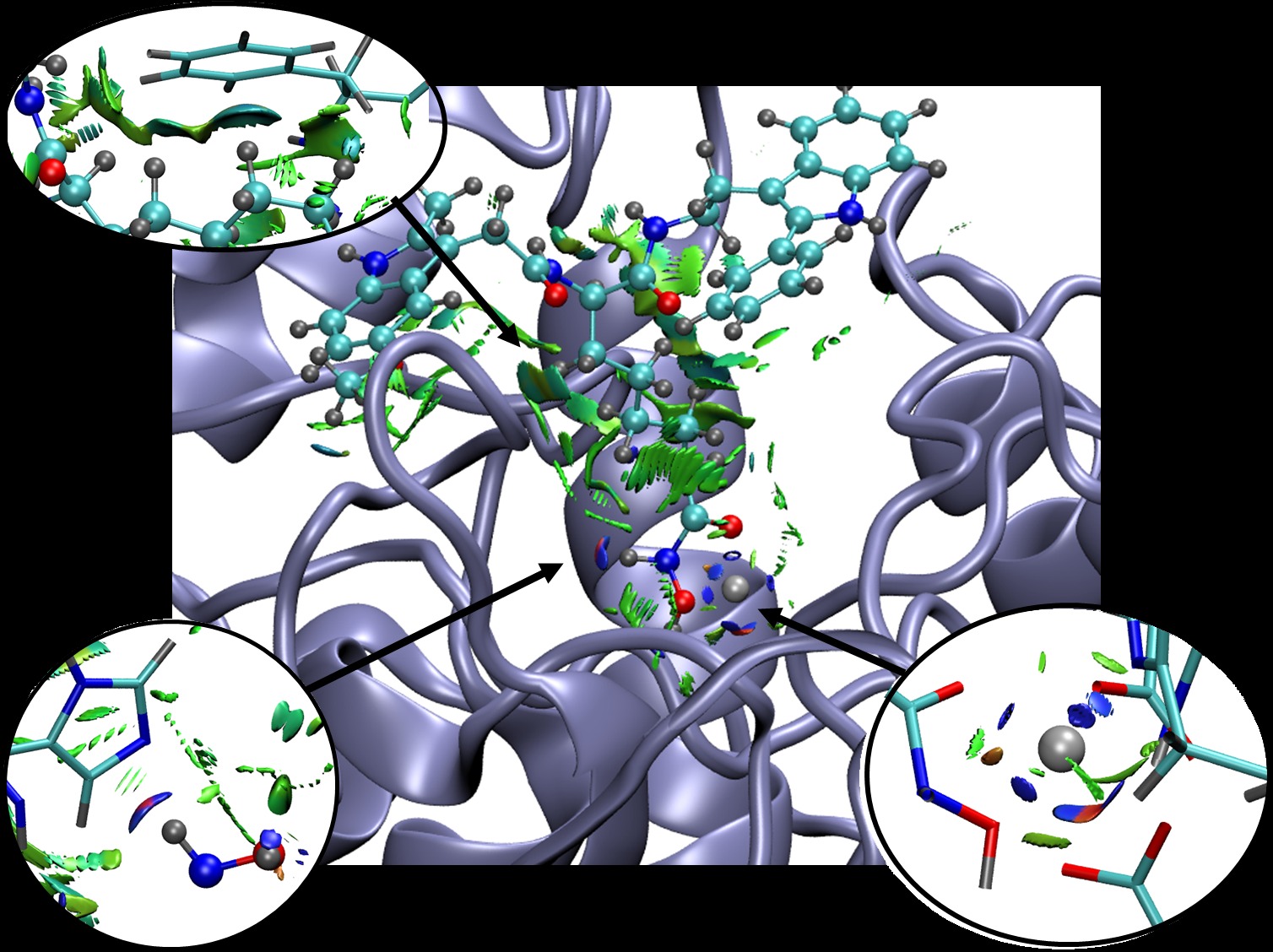
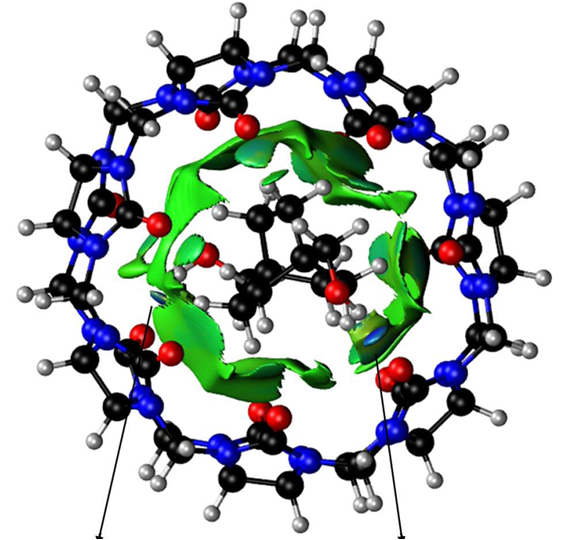
This code enables graphical visualization of inter and intramolecular non-covalent interactions (i.e. hydrogen bonds, steric clashes and van der Waals) in systems ranging from small molecules to large biosystems.
NCI (Non-Covalent Interactions) is a visualization index based on the electron density and its derivatives. It enables identification of non-covalent interactions, based on the peaks that appear in the reduced density gradient (RDG) at low densities. RDG isosurfaces for these peaks enable the visualization of weak interactions. The isosurfaces correspond to both favorable and unfavorable interactions, as differentiated by the sign of the second density Hessian eigenvalue. The sign of this eigenvalue, times the density, is able to characterize both the strength and (un)favourable nature of the interactions and defines the isosurface colouring. See the reference below for a thorough explanation of the physics.
This program computes density and reduced density gradient (RDG) on a grid and provides Gaussian-format cube files and VMD scripts for the direct visualization of the results. It can be run using either SCF densities (wfn input files) or promolecular densities (xyz input files), which makes it applicable to large biosystems.
Want to learn how to use NCIPlot through examples?
|
|
|
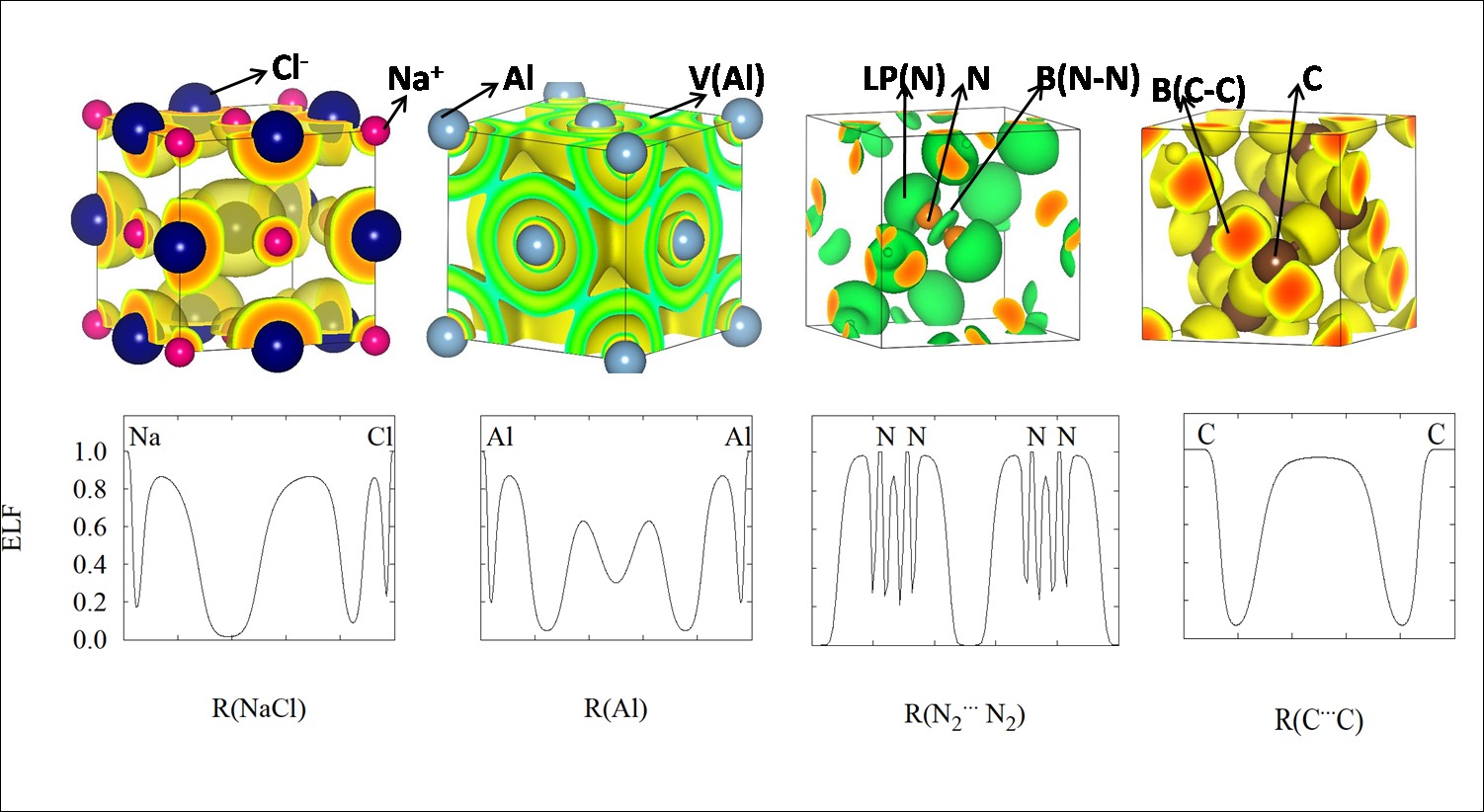
This programs is based on a novel computational procedure, general, automated and robust, for the analysis of local and global properties of the electron localization function (ELF) in crystalline solids.
The algorithm successfully faces the two main shortcomings of the ELF analysis in crystals:
|