Next: 36.2.2 Defining active geometry
Up: 36.2 Automatic geometry optimization
Previous: 36.2 Automatic geometry optimization
36.2.1 Optimization coordinates (COORD)
It is possible to use various coordinate types and algorithms for the optimization. This can
be controlled by additional subcommands as described in this and the following subsections.
COORD,[opt_space],[opt_coord],[displacement_type],[option]
This option chooses the optimization space and the coordinate system in which the optimization takes place.
opt_space defines the parameters to be optimized. By default, if the geometry input
is given in z-matrix format, all variables on which the z-matrix depends are optimized.
Subsets of the variables on which the z-matrix depends can be chosen using the
ACTIVE or INACTIVE subdirectives.
If the z-matrix depends on no variables or xyz input is used, all 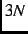 cartesian
coordinates are optimized.
cartesian
coordinates are optimized.
opt_coord determines the coordinates in which the optization takes place. By
default, local normal coordinates are used. Optionally cartesian coordinates or natural internal
coordinates can be used.
displacement_type specifies how numerical gradients and hessians are computed.
This defaults to symmetric displacement coordinates and should normally not be modified.
These defaults can be modified using the COORD directive.
opt_space can be one of the following:
- ZMAT
- Optimize all variables on which the z-matrix depends (default)
- 3N
- Optimze all cartesian coordinates.
Z-Matrix input coordinates will be destroyed on this entry.
opt_coord can be one of the following:
- NORMAL
- Optimization in local normal coordinates.
This is default if the Model Hessian is used to approximate the hessian.
- NONORM
- Don't use local normal coordinates.
- BMAT[=filename]
- Use Pulay's natural internal coordinates,
see G. Fogarasi, X. Zhou, P. W. Taylor and P. Pulay
J. Am. Chem. Soc. 114, 8191 (1992); P. Pulay, G. Fogarasi, F. Pang, J. E. Boggs
J. Am. Chem. Soc. 101, 2550 (1979)).
Optionally, the created coordinates plus additional information about this optimization is written
to the specified file.
These coordinates resemble in part the valence coordinates used by vibrational spectroscopists,
and have the advantage of decreasing coupling between different modes. This often
increases the speed of convergence. The use of this option is highly recommended,
especially in minimization of large organic molecules with rings.
Nevertheless you should keep in mind that these coordinates are constructed
automatically, and there exist exotic bond structures which might not be
treated properly (e.g. weakly bonded species as in
transition state optimizations). In such a case, if the BMAT
optimization converges slowly or leads to symmetry-breaking errors,
you should try another optimization method and/or cartesian or Z-Matrix coordinates.
displacement_type can be one of the following (affects only numerical gradients):
- SYM
- Use symmetric displacement coordinates (default). This is the
only recommended option.
- CART
- Use cartesian displacements (not recommended). This requires many more energy
calculations than necessary and does not preserve the molecular symmetry.
- UNIQUE
- Use symmetry-unique cartesian displacements (not recommended)
If option is set to [NOROT], the cartesian coordinates are not transformed to minimze rotations.
Next: 36.2.2 Defining active geometry
Up: 36.2 Automatic geometry optimization
Previous: 36.2 Automatic geometry optimization
molpro@molpro.net
Feb 26, 2003