For all atomic integrals, the proper expression for the integral is
given, together with the labels written on the file
AOPROPER , for
reference in later stages of a DALTON calculation (like for instance
in during the evaluation of dynamic response properties, or for
non-DALTON programs).
- .ANGLON
- Contribution to the one-electron contribution of
the magnetic moment using London
orbitalsindexLondon orbitals arising from the differentiation on
the London phase factors, see Ref. [68].
- Integral:
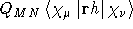
- Property label:
XANGLON , YANGLON , ZANGLON
- .ANGMOM
- Angular momentum around the molecular origin.
This can be adjusted by changing the gauge origin through the use of
the .GAUGEO
keyword.
- Integral:
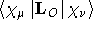
- Property label:
XANGMOM , YANGMOM , ZANGMOM
- .CARMOM
READ (LUCMD,*) IORCAR
Cartesian multipole integrals to order
IORCAR. Read one more
line specifying order. See also the keyword .SPHMOM
.
- Integral:
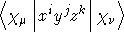
- Property label:
CMiijjkk
where i+j+k =IORDER.
- .CM-1
READ (LUCMD, '(A7)') FIELD1
First derivative of the electric dipole operator with respect to an
external magnetic field due to differentiation of the London phase
factors, see Ref. [54]. Read one more line giving
the direction of the electric field (A7) . These
include X-FIELD, Y-FIELD, and Z-FIELD.
- Integral:
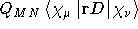
- Property label:
D-CM1 X , D-CM1 Y , D-CM1 Z
where D is the direction of the applied electric field as specified in
the input.
- .CM-2
- Second derivative electric dipole operator
with respect to an external magnetic field due
to differentiation of
the London phase factors, see Ref. [54]. Read one
more line giving the direction of the electric
field (A7) . These
include
X-FIELD, Y-FIELD, and Z-FIELD.
- Integral:
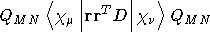
- Property label:
D-CM2 XX, D-CM2 XY, D-CM2 XZ,
D-CM2 YY, D-CM2 YZ, D-CM2 ZZ
where D is the direction of the applied electric field as specified in
the input.
- .DARWIN
- One-electron Darwin integrals .
- Integral:
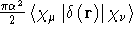
- Property label:
DARWIN
- .DIASUS
- Diamagnetic magnetizability integrals, as
calculated with London atomic orbitals, see Ref. [68].
- Integral:
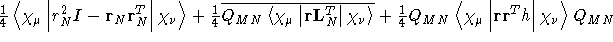
- Property label:
XXdh/dB2, XYdh/dB2,
XZdh/dB2, YYdh/dB2, YZdh/dB2, ZZdh/dB2
- .DIPLEN
- Dipole length integrals.
- Integral:
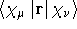
- Property label:
XDIPLEN , YDIPLEN , ZDIPLEN
- .DIPORG
READ (LUCMD, *) (DIPORG(I), I = 1, 3)
Specify the dipole origin to be used in the
calculation. Read one more
line containing the three Cartesian components (*). Default is (0,0,0).
- .DIPVEL
- Dipole velocity integrals.
- Integral:
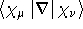
- Property label:
XDIPVEL , YDIPVEL , ZDIPVEL
- .DSO
- Diamagnetic spin-orbitindexdiamagnetic spin-orbit
integrals. These are
calculated using Gaussian quadrature as
described in
Ref. [69]. The number of quadrature point is controled by
the keyword .POINTS
.
- Integral:
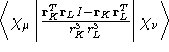
- Property label:
DSO abcd where ab is the symmetry
coordinate of a given component for the symmetry-adapted nucleus K,
and cd is in a similar fashion the symmetry coordinate for the
symmetry-adapted nucleus L.
- .DSUSLH
- The contribution to diamagnetic
magnetizability
integrals from the differentiation of the London orbital
phase-factors, see Ref. [68].
- Integral:
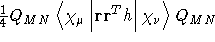
- Property label:
XXDSUSLH, XYDSUSLH,
XZDSUSLH, YYDSUSLH, YZDSUSLH, ZZDSUSLH
- .DSUSLL
- The contribution to the diamagnetic
magnetizability integrals from mixed differentiation on the Hamiltonian
and the London orbital phase factors , see Ref. [68].
- Integral:
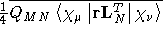
- Property label:
XXDSUSLL, XYDSUSLL,
XZDSUSLL, YYDSUSLL, YZDSUSLL, ZZDSUSLL
- .DSUSNL
- The contribution to the diamagnetic
magnetizability integrals using
London orbitals but with
contributions from the differentiation of the Hamiltonian only, see
Ref. [68].
- Integral:
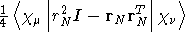
- Property label:
XXDSUSNL, XYDSUSNL,
XZDSUSNL, YYDSUSNL, YZDSUSNL, ZZDSUSNL
- .DSUTST
- Test of the diamagnetic magnetizability
integrals with London atomic
orbitals . Mainly for debugging purposes.
- .EFGCAR
- Cartesian electric field gradient
integrals .
- Integral:
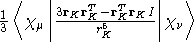
- Property label:
xyEFGabc, where x and y are
the cartesian directions, ab the number of the symmetry
independent center, and c that centers c'th symmetry-generated
atom.
- .EFGSPH
- Spherical electric field gradient
integrals. Obtained by transforming the Cartesian electric-field
gradient integrals (see .EFGCAR
) to spherical basis.
- .EXPIKR
READ (LUCMD, *) (EXPKR(I), I = 1, 3)
Cosine and sine integrals.
Read one more line containing the wave numbers in the three Cartesian
directions. The center of expansion is always (0,0,0).
- .FC
- Fermi-contact integrals, see
Ref. [33].
- Integral:
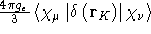
- Property label:
FC NAMab, where NAM is the three
first letters in the name of this atom, as given in the
MOLECULE.INP file, and ab is the number of the
symmetry-adapted nucleus.
- .GAUGEO
READ (LUCMD, *) (GAGORG(I), I = 1, 3)
Specify the gauge origin to be used in the
calculation. Read one more line containing the three Cartesian
components (*). Default is (0,0,0).
- .HBDO
- Geometric half-differentiated overlap
matrix
differentiated once more with respect to magnetic field as given in Eq. (59)
in Ref. [48].
- .HDO
- Symmetrized, half-differentiated
overlap integrals with respect to geometric distortions , see
Ref. [70]. Differentiation on the ket-vector.
- Integral:
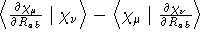
- Property label:
HDO ab , where ab is the number
of the symmetry-adapted coordinate being differentiated.
- .HDOBR
- Geometric half-differentiated overlap
matrix
differentiated once more on the ket-vector with respect to external
magnetic field as given in Eq. (60) of Ref. [48].
- .HDOBRT
- Test the calculation of the .HDOBR
integral. Mainly for debugging purposes.
- .INPTES
- Test the correctness of the **INTEGRALS
-input. Mainly
for debugging purposes, but also a good option to check if the MOLECULE input
has been typed in correctly.
- .KINENE
- Kinetic energy integrals . Note however, that the kinetic energy integrals used in the
wave function optimization is generated in the *ONEINT
section.
- Integral:

- Property label:
KINENERG, where ab is the number
of the symmetry-adapted nuclear coordinate.
- .LONMOM
- Contribution to the London magnetic
moment from
the differentiation with respect to magnetic field on the London
orbital phase factors, see Ref. [68].
- Integral:
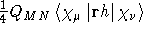
- Property label:
XLONMOM , YLONMOM , ZLONMOM .
- .MAGMOM
- One-electron contribution to the magnetic
moment
around the nuclei to which the
atomic orbitals are attached. This is the London atomic
orbital
magnetic moment as defined in Eq. (35) of
Ref. [31].
- Integral:
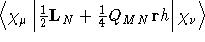
- Property label:
dh/dBX , dh/dBY , dh/dBZ .
- .MASSVE
- Mass-velocity integrals.
- Integral:

- Property label:
MASSVELO, where ab is the number
of the symmetry-adapted nuclear coordinate.
- .MGMO2T
- Test of two-electron integral contribution to
magnetic moment.
- .MGMOMT
- Test the calculation of the .MAGMOM
integrals.
- .MGMTHR
READ (LUCMD, *) PRTHRS
Set the threshold for which two-electron integrals should be tested
with the keyword .MGMO2T
. Default is 10 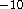 .
.
- .NELFLD
- Nuclear electric field integrals .
- Integral:
 where K is the nucleus of interest.
where K is the nucleus of interest. - Property label:
NEF ab , where ab is the number
of the symmetry-adapted nuclear coordinate.
- .NO HAM
- Do not calculate ordinary Hamiltonian integrals.
- .NOSUP
- Do not calculate the supermatrix
integral file.
This may be required in order to reduce the amount of disc space used
in the calculation (to approximately one-third before entering the
evaluation of molecular properties).
Note however, that this will increase the time used for the evaluation
of the wave function
significantly in ordinary Hartree-Fock runs. It is default for direct and
parallel calculations.
- .NOTWO
- Only calculate the one-electron part of the
Hamiltonian integrals. It is default for direct and parallel calculations.
- .NPOTST
- Test of the nuclear potential integrals
calculated with the keyword .NUCPOT
. Mainly for debugging
purposes.
- .NSLTST
- Test of the integrals calculated with the
keyword .NSTLON
. Mainly for debugging purposes.
- .NSNLTS
- Test of the integrals calculated with the
keyword .NSTNOL
. Mainly for debugging purposes.
- .NST
- Calculate the one-electron contribution to the
diamagnetic nuclear shielding
tensor integrals using London atomic
orbitals , see Ref. [68].
- Integral:
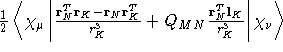 where K is the nucleus of interest.
where K is the nucleus of interest. - Property label:
NST ab c, where ab is the number
of the symmetry-adapted nuclear magnetic moment coordinate, and
c refers to the x, y, or z component of the magnetic field.
- .NSTCGO
- Calculate the diamagnetic nuclear shielding
ytensor integrals without using London atomic orbitals . Note that the
gauge origin is controled by the keyword .GAUGEO
.
- Integral:
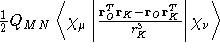 where K is the nucleus of interest.
where K is the nucleus of interest. - Property label:
NSCOab c, where ab is the number
of the symmetry-adapted nuclear magnetic moment coordinate, and
c refers to the x, y, or z component of the magnetic field. O
is the gauge origin.
- .NSTLON
- Calculate the contribution to the nuclear
shielding tensor from the differentiation of the London orbital
phase-factors , see Ref. [68].
- Integral:
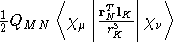 where K is the nucleus of interest.
where K is the nucleus of interest. - Property label:
NSLOab c, where ab is the number
of the symmetry-adapted nuclear magnetic moment coordinate, and
c refers to the x, y, or z component of the magnetic field.
- .NSTNOL
- Calculate the contribution to the nuclear
shielding tensor from the differentiation of the Hamiltonian alone , see Ref. [68].
- Integral:
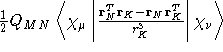 where K is the nucleus of interest.
where K is the nucleus of interest. - Property label:
NSNLab c, where ab is the number
of the symmetry-adapted nuclear magnetic moment coordinate, and
c refers to the x, y, or z component of the magnetic field.
- .NSTTST
- Test the calculation of the one-electron
diamagnetic nuclear shielding
tensor using London atomic orbitals .
- .NUCMOD
READ (LUCMD, *) INUC
Choose nuclear model. A 1 corresponds to a point nucleus (which is the
default), and 2 corresponds to a Gaussian distribution model.
- .NUCPOT
- Calculate the nuclear potential energy.
Currently this keyword can only be used in calculations not employing
symmetry.
- Integral:
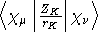 where K is the nucleus of interest.
where K is the nucleus of interest. - Property label:
POT.E ab, where ab is the two
first letters in the name of this nuclei. Thus note that in order to
distinguish between integrals, the fisrt to letters in an atoms name
must be unique.
- .PHASEO
READ (LUCMD, *) (ORIGIN(I), I = 1, 3)
Set the origin appearing in the London atomic orbital phase-factors.
Read one more line containing the Cartesian components of this origin (*).
Default is (0,0,0).
- .POINTS
READ (LUCMD,*) NPQUAD
Read the number of quadrature points to be
used in the evaluation of
the diamagnetic spin-orbit integrals, as
requested by the keyword
.DSO
. Read one more line containing the number of quadrature
points. Default is 40.
- .PRINT
READ (LUCMD,*) IPRDEF
Set default print level during the integral evaluation. Read one more line
containing print level. Default is the value of IPRDEF
from the general input module for DALTON .
- .PROPRI
- Print all one-electron property integrals requested.
- .PSO
- Paramagnetic spin-orbit integrals , see
Ref. [33].
- Integral:
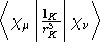 where K is the nucleus of interest.
where K is the nucleus of interest. - Property label:
PSO ab , where ab is the number
of the symmetry-adapted nuclear magnetic moment coordinate.
- .QUADRU
- Quadrupole moment integrals. For traceless quadrupole moment integrals as
defined by Buckingham [23], see the keyword .THETA
.
- Integral:
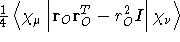
- Property label:
XXQUADRU, XYQUADRU,
XZQUADRU, YYQUADRU, YZQUADRU, ZZQUADRU
- .QUASUM
- Calculate all atomic integrals as square
matrices, irrespective of their inherent Hermiticity or
anti-Hermiticity.
- .S1MAG
- Calculate the first derivative overlap
matrix with respect to an
external magnetic field by differentiation
of the London phase factors , see Ref. [68].
- Integral:

- Property label:
dS/dBX , dS/dBY ,
dS/dBZ
- .S1MAGL
- Calculate the first magnetic half-differentiated overlap
matrix with respect to an
external magnetic field as needed with the
natural connection , see
Ref. [32]. Differentiated
on the bra-vector.
- Integral:
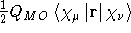
- Property label:
d<S/dBX|, d<S/dBY|,
d<S/dBZ|
- .S1MAGR
- Calculate the first magnetic half-differentiated overlap
matrix with respect to an external magnetic field as needed with the
natural connection , see Ref. [32]. Differentiated on
the ket-vector.
- Integral:
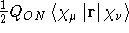
- Property label:
dS>/dBX|, dS>/dBZ|,
dS>/dBZ|
- .S1MAGT
- Test the integrals calculated with the keyword
.S1MAG
. Mainly for debugging purposes.
- .S1MLT
- Test the integrals calculated with the keyword
.S1MAGL
. Mainly for debugging purposes.
- .S1MRT
- Test the integrals calculated with the keyword
.S1MAGR
. Mainly for debugging purposes.
- .S2MAG
- Calculate the second derivate of the overlap
matrix with respect to an external magnetic field by differentiation
of the London phase factors , see Ref. [68].
- Integral:
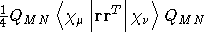
- Property label:
dS/dB2XX, dS/dB2XY,
dS/dB2XZ, dS/dB2YY, dS/dB2YZ, dS/dB2ZZ
- .S2MAGT
- Test the integrals calculated with the keyword
.S2MAG
. Mainly for debugging purposes.
- .SD
- Spin-dipole integrals , see
Ref. [33].
- Integral:
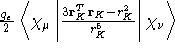
- Property label:
SD ab c, where ab is the number
of the first symmetry-adapted coordinate (corresponding to
symmetry-adapted nuclear magnetic moments) and c is the x, y,
or z component of the magnetic moment with respect to spin coordinates.
- .SD+FC
- Calculate the sum of the spin-dipole and
Fermi-contact integrals .
- Integral:
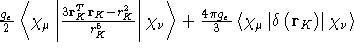
- Property label:
SDC ab c, where ab is the number
of the first symmetry-adapted coordinate (corresponding to
symmetry-adapted nuclear magnetic moments) and c is the x, y,
or z component of the magnetic moment with respect to spin coordinates.
- .SECMOM
- Second-moment integrals .
- Integral:
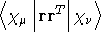
- Property label:
XXSECMOM, XYSECMOM,
XZSECMOM, YYSECMOM, YZSECMOM, ZZSECMOM
- .SELECT
READ (LUCMD, *) NPATOM
READ (LUCMD, *) (IPATOM(I), I = 1, NPATOM
Select which atoms for which a given atomic integral is to be
calculated. This applies mainly to property integrals for which there
exist a set of integrals for each nuclei. Read one more line
containing the number of atoms selected, and then another line
containing the numbers of the atoms selected. Most useful when
calculating diamagnetic spin-orbit
integrals, as this is a rather
time-consuming calculation. The numbering is of symmetry-independent
nuclei.
- .SORT I
- Requests that the
two-electron integrals should be
sorted for later use in SIRIUS . See also keywords .PRESORT
in the
**DALTON
and *TRANSFORMATION
input sections.
- .SOTEST
- Test the calculation of spin-orbit integrals as
requested by the keyword .SPIN-O
.
- .SPHMOM
READ (LUCMD,*) IORSPH
Spherical multipole integrals to order
IORSPH. Read one more
line specifying order. See also the keyword .CARMOM
.
where i+j+k =IORDER.
- .SPIN-O
- Spatial spin-orbit
integrals, see Ref. [71]. Both the one- and the
two-electron integrals are calculated, the latter stored on the file
AO2SOINT.
- One-electron Integral:
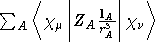 where
where 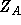 is the charge of nucleus A and the summation runs over
all nuclei of the molecule.
is the charge of nucleus A and the summation runs over
all nuclei of the molecule. - Property label:
X1SPNORB, Y1SPNORB, Z1SPNORB - Two-electron Integral:
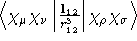
- Property label:
X2SPNORB, Y2SPNORB, Z2SPNORB
- .SQHDOR
- Square, non-symmetrized half differentiated
overlap integrals with respect to geometric distortions , see
Ref. [70]. Differentiation on the ket-vector.
- Integral:
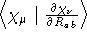
- Property label:
SQHDORab, where ab is the number
of the symmetry-adapted coordinate being differentiated.
- .SUPONL
- Only calculate the supermatrix. Requires the
presence of the two-electron integral file .
- .SUSCGO
- Diamagnetic magnetizability integrals calculated
without the use of London atomic orbitals. The choice of gauge
origin
can be controled by the keyword .GAUGEO
.
- Integral:
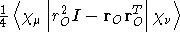
- Property label:
XXSUSCGO, XYSUSCGO,
XZSUSCGO, YYSUSCGO, YZSUSCGO, ZZSUSCGO
- .THETA
- Traceless quadrupole moment integrals as defined by Buckingham [23].
- Integral:
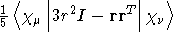
- Property label:
XXTHETA , XYTHETA ,
XZTHETA , YYTHETA , YZTHETA , ZZTHETA