



Next: Getting the wave function
Up: Getting started with DALTON
Previous: The MOLECULE input file
If we have made two input files, one corresponding to
DALTON.INP and one corresponding to the MOLECULE \
input file, we
are ready to do our first calculation. Examples of input files can be
found in the dalton/test directory in order to give you a
head-on start with DALTON .
Calculations with DALTON is most conveniently done using the
supplied shell-script dalton. Thus,
to run a calculation of
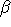 (first hyperpolarizability)
with input available as
(first hyperpolarizability)
with input available as
beta.dal on the HCl molecule, with molecule input available as
hcl.mol. In order to run this calculation you would type
> dalton beta hcl
assuming dalton is available in your path. When the job is
finished, the output is copied back as beta_hcl.out. In case that the
dalton- and molecule-input has the same name (e.g. dalton test1 test1), the output will be named test1.out.
There are several options to this script, which can be viewed by
typing dalton -h or just dalton. These options include:
- -a
- Copy various auxillary files from the
home directory to the
scratch directory before the calculation starts. This is needed in
case of restarts of intrisic reaction coordinate, direct dynamics,
Raman Optical Activity, or general numerical differentiation, where
information stored in the files
DALTON.IRC, DALTON.TRJ,
and DALTON.WLK respectively is needed.
- -A
- Copy various auxillary files from the scratch directory to the home
directory after the calculation has completed. These auxillary files
contains extra information that may become useful for certain kinds of
calculations. All these auxillary files are described below.
- -b directory
- Give an alternative directory where the program
should look for basis sets . Needed in case you want to use local
modifications of a given basis. Note that the directory must end with
a
/.
- -d
- Removes the contents of the scratch directory before a
calculation starts in order to avoid inconsistencies between files.
- -D
- Do not remove the content of the scratch directory. By
default the scratch directory will be deleted. However, in order to do
a restart you will need to keep all files in this directory, and you
then need to add the -D option when submitting the job.
- -e ext
- Change the extension to the output file from
.out
to .ext.
- -f
- Copy the fort.21 file from your home directory to the scratch
directory before the calculation starts. This is needed in order to be
able to do a restart in the wave function optimization.
- -F
- Copy the fort.21 file from the scratch
directory to your home
directory after the calculation has finished. This is needed if you
want to (re)start from the wave function created in this run.
- -m mem
- Change the default size of the scratch
memory space to
mem words.
- -n nodes
- Number of nodes to be used in a parallel MPI calculation
controlled by running mpirun.
- -t tmpdir
- Change the
TMPDIR (the scratch disc area) to
tmpdir from the default scratch directory determined at
install-time.
In most cases, the DALTON.OUT file will contain all the
information needed about a given calculations. However, in certain
cases, additional information may be wanted, and this is contained in
various sets of auxillary files. Their contents is described briefly
here. We note that in order for these files to be copied back to your
job directory the flag -a must be supplied to the dalton
run script.
- DALTON.BAS
- Contains a dump of a complete molecule input file
obtainable by the keyword .MOLPRI
. This file take maximum
advantage of formatted input, yet differences may occur compared to
the basis sets obtained from the basis set library due to the
restricted number of digits available in the standard-form output.
- DALTON.MOL
- Contains the information needed by the
MOLPLT-program for visualizing the molecular geometry. The
MOLPLT-program is distributed with the GAMESS-US program
package .
- DALTON.NCA
- Contains the information needed by the
MOLPLT-program for visualizing normal coordinates .
- DALTON.ORB
- Contains information about basis set and
MO-coefficients so that MO density plots may be generated using the
PLTORB program that comes with the GAMESS-US
distribution. Currently not supported .
- DALTON.IRC
- Contains information obtained from an Intrinsic
reaction Coordinate (IRC) calculation, as described in
Sec.
 .
.
- DALTON.WLK
- Contains information from the walk-procedure, and is
needed when restarting a walk (e.g. a numerical differentiation).
- DALTON.TRJ
- Contains information from a direct dynamics
calculation as described in Sec. .
- DALTON.CM
- An output file that contains the most essential
information needed for calculation of shielding and magnetizability
polarizabilities. Most easily used together with the analyzing program
ODCPRG.f supplied in the tool-directory .
- DALTON.HES
- If the keyword .HESPUN
has been specified in
the *VIBANA
input module, the molecular Hessian
will be written
to this file in a standard format, which may be used as a start Hessian
in an first-order geometry
optimization, or as input to a ROA or VCD
analysis with different basis sets/level of correlation for the
intensity operators and the force field. See also the
FChk2HES.f program in the tool-directory.
- DALTON.MOPUN
- Contains the molecular-orbital coefficients
printed in a standard format allowing the transfer of molecular
orbitals coefficients from one computer to another.
Next: Getting the wave function
Up: Getting started with DALTON
Previous: The MOLECULE input file
Kenneth Ruud
Sat Apr 5 10:26:29 MET DST 1997