



Next: Z-matrix input
Up: MOLECULE input style
Previous: General MOLECULE input
Assuming that we have given the general input as indicated above, we
now want to specify the spatial arrangements of the atoms in a
Cartesian coordinate system. We will also for sake of illustration
assume that we have given explicitly the generators of the point group
to be used in the calculation (in this case C 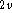 , with the yz- and
xz-planes as mirror planes).
, with the yz- and
xz-planes as mirror planes).
In tetrahedran we will have two different kinds of atoms, carbon and
hydrogen, as indicated by the number 2 on the fourth (fifth) line of
the input. We will also assume that we punch the basis set ourselves,
in order to present the input format for the basis set.
For tetrahedran, the input would then look like
5: 6. 2 3 1 1 1
6:C1 1.379495419 .0 0.975450565
7:C2 .0 1.379495419 -.975450565
8: 8 3
9:486.9669 .01772582
10:73.37109 .1234779
11:16.41346 .4338754
12:4.344984 .5615042
13:8.673525 -.1213837
14:2.096619 -.2273385
15:.6046513 1.185174
16:.1835578 1.00000
17: 4 2
18:8.673525 .06354538
19:2.096619 .2982678
20:.6046513 .7621032
21:.1835578 1.000000
22: 1 1
23:0.8 1.0
24: 1. 2 2 1 1
25:H1 3.020386510 .0 2.1357357837
26:H2 .0 3.020386510 -2.1357357837
27: 4 2
28:18.73113 .03349460
29:2.825394 .2347270
30:.6401218 .8137573
31:.1612778 1.000000
32: 1 1
33:0.75 1.0
The different lines are:
- 5
-
ZMATL, CHARGE, NUMBER, MAXL, NGRP(I)
(BN,A6,F4.0,24I5).
- ZMATL
- This field must be left blank when Cartesian
coordinate input is used as the field
is used for checking whether
Cartesian coordinates or Z-matrix input is being
used. Characters
placed in this field will tell the program to use Z-matrix input.
- CHARGE
- Charge of this atom type.
- NUMBER
- Number of symmetry-distinct atoms of
this type (or, if the symmetry detection routines are being used, all
atoms of this kind).
- MAXL
- Maximum angular quantum number used in the
basis set for this atom type (s=1, p=2, etc.). Not required if the
basis set library is being used.
- NGRP(I)
- Number of groups (blocks) of generally contracted
functions of angular quantum number I. The loop is over MAXL. Not
required if the basis set library is used. It is noteworthy that
DALTON collects all basis functions into one such shell, and
evaluates all integrals arising from that shell simultaneously, and
the memory requirements grow rapidly with the number of basis
functions in a shell (note for instance that four g functions
actually are 36
basis functions, as there are 9 components of each g
function). Memory requirements can therefore be reduced
by splitting
basis functions of the quantum number into different blocks. However,
this will decrease the performance of the integral calculation.
- 6
-
NAME, X, Y, Z, FLAG (A4,3F20.15)
- NAME
- Atom name. A different name should be used for
each atom of the same type, although this is not required.
- X
- x-coordinate (in atomic units, unless Ångström
has been requested on line 4 of the input).
- Y
- y-coordinate.
- Z
- z-coordinate.
The Cartesian coordinates may
be given in free format. However, the name of the atom must still be
left four places, and no coordinates must enter the four first
positions.
- 7
- This is the other symmetry-distinct center of this type.
- 8
-
FRMT, NPRIM, NCONT, NOINT (A1,I4,2I5).
- FRMT
- A single character describing the input format of the
basis set in this block. The default format is (8F10.4) which
will be used if FRMT is left blank. In this format
the first column is the orbital exponent and the seven last columns
are contraction coefficients. If no numbers are given, a zero is
assumed. If more than 7 contracted functions occur in a given block,
the contraction coefficients may be continued on the next line, but
the first column (where the orbital exponents are given) must then be
left blank.
An F or f in the first
position will indicate that the input is in free format. This will of
course require that all contraction coefficients need to be typed in,
as all numbers need to be present on each line. However, note that
this options is particularly handy together with completely
decontracted basis sets, as described below. Note that the program
reads the free format input from an internal file that is 80 charcters
long, and no line can therefore exceed 80 characters.
One may also give the format H or h. This corresponds to
high precision format (4F20.8), where the first column again is
reserved for the orbital exponents, and the three next lines are
designated to the contraction coefficients. If no number is given, a zero
is assumed. If there are more than three contracted orbitals in a
given block, the contraction coefficients may be continued on the next
line, though keeping the column of the orbital exponents blank.
- NPRIM
- Number of primitive
Gaussians in this block.
- NCONT
- Number of contracted
Gaussians in this block. If a zero
is given, an uncontracted basis set will be assumed, and only orbital
exponents need to be given.
- 9
-
EXP, (CONT(I), I=1,NCONT)
- EXP
- Exponent of this primitive.
- CONT(I)
- Coefficient of this primitive in contracted
function I.
We note that the format of the orbital exponents and the contraction
coefficients are determined from the value of FRMT defined on
line 8.
- 10-16
- These lines complete the specification of this
contraction block: the s basis here.
- 17-21
- New contraction block (see lines 8 and 9 above).
- 22-23
- New contraction block.
- 24-33
- Specifies a new atom type: coordinates and basis set.
Next: Z-matrix input
Up: MOLECULE input style
Previous: General MOLECULE input
Kenneth Ruud
Sat Apr 5 10:26:29 MET DST 1997