



Next: Coupled-cluster calculations, CC
Up: Electron Spin Resonance: *ESR
Previous: Zero-field splitting: .ZFS
Contents
Index
Electronic g-tensors: .G-TENSOR
Calculation of the electronic g-tensor:
- .G-TENSOR
Initializes input block for g-tensor related options
The default is to calculate all contributions. The following
options selects individual contributions
- .RMC
Relativistic mass correction
- .OZSO1
Second-order (paramagnetic) orbital-Zeeman + 1-electron spin-orbit contributions
- .OZSO2
Second-order (paramagnetic) orbital-Zeeman + 2-electron spin-orbit contributions
- .GC1
1-electron gauge correction (diamagnetic) contributions
- .GC2
2-electron gauge correction (diamagnetic) contributions
- .ECC
Choose electron center of charge (ECC) as gauge origin.
The following are utility options for modifying the default calculational
procedure.
- .ADD-SO
Adds the 1- and 2-electron spin-orbit operators. This
option may be used when one is not interested in the individual
1- and 2-electron contribution to the paramagnetic g-tensor,
since it reduces the number of response equations to be solved.
- .MEAN-FIELD
Uses the approximate atomic mean field (AMFI) spin-orbit operator
for evaluating the paramagnetic contributions.
- .SCALED
Uses the approximate 1-electron spin-orbit operator with scaled nuclear
charges taken from Ref. [172] for evaluating the paramagnetic contributions.
- .ZERO
READ(LUCMD,'(A80)')G_LINE
This option is mainly for linear molecules in a 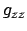 state.
Specifying e.g. "ZZ" on the input line instructs the program
to skip the calculation of the paramagnetic contribution to
state.
Specifying e.g. "ZZ" on the input line instructs the program
to skip the calculation of the paramagnetic contribution to 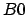 .
.
Next: Coupled-cluster calculations, CC
Up: Electron Spin Resonance: *ESR
Previous: Zero-field splitting: .ZFS
Contents
Index
Dalton Manual - Release 1.2.1