



Next: *ORBITAL INPUT
Up: Main input groups in
Previous: *NEVPT2 INPUT
Contents
Index
*OPTIMIZATION
Purpose:
To change defaults for optimization of an MCSCF wave function.
Some of the options also affect a QC-HF optimization.
- .ABSORPTION
- READ (LUINP,'(A8)') RWORD
RWORD = ` LEVEL 1', ` LEVEL 2', or ` LEVEL 3'
Orbital absorption in MCSCF optimization
at level 1, 2, or 3, as specified
(normally level 3, see comments below). This keyword may be repeated to
specify more than one absorption level, the program will then begin with
the lowest level requested and, when that level is converged,
disable the lower level and shift to the next level.
- .ACTROT
- include specified active-active rotations
READ (LUINP,*) NWOPT
DO I = 1,NWOPT
READ (LUINP,*) JWOP(1,I),JWOP(2,I)
END DO
JWOP(1:2,I) denotes normal molecular orbital numbers (not the active
orbital numbers).
- .ALWAYS ABSORPTION
- Absorption in all MCSCF macro iterations
(default is to disable absorption in
local region or after ".MAXABS" macro iterations, whichever comes first).
Absorption is always disabled after Newton-Raphson algorithm has been used,
and thus also when doing ".CORERELAX",
because absorption may cause variational collapse if the desired state is excited.
- .CI PHP MATRIX
- Default : MAXPHP = 1 (Davidson's algorithm)
READ (LUINP,*) MAXPHP
PHP is a subblock of the CI matrix which is calculated explicitly
in order to obtain improved CI trial vectors compared to the
straight Davidson algorithm. The
configurations corresponding to
the lowest diagonal elements are selected, unless
".PHPRESIDUAL" is specified.
MAXPHP is the maximum dimension of PHP, the actual dimension
will be less if MAXPHP will split degenerate configurations.
- .COREHOLE
- READ (LUINP,*) JCHSYM,JCHORB
JCHSYM = symmetry of core orbital
JCHORB = the orbital in symmetry JCHSYM with a single core hole
Single core hole MCSCF calculation. The calculation must be of RAS type
with only the single core-hole orbital in RAS1, the state specified with
".STATE" is optimized with the core-hole orbital
frozen.
The specified core hole orbital must be either inactive or
the one RAS1 orbital, if it is inactive then it will switch places with
the RAS1 orbital and it will not be possible to also
specify ".REORDER". If explicit reordering is required you must reorder
the core orbital yourself and let JCHORB point to the one RAS1 orbital.
Orbital absorption is activated at level 2. See comments below for more information.
- .CORERELAX
- (ignored if ".COREHOLE" isn't also specified)
Optimize state with relaxed core orbital (using Newton-Raphson algorithm,
it is not necessary to explicitly specify ".NR ALWAYS").
It is assumed that this calculation follows an optimization
with frozen core orbital and that the orbital has already been
moved to the RAS1 space (i.e., the specific value of
"JCHORB" under ".COREHOLE" is ignored). Any
orbital absorption will be ignored.
- .DETERMINANTS
- Use determinant basis instead of CSF basis (see comments).
- .EXACTDIAGONAL
- Default for RAS calculations.
Use the exact orbital Hessian diagonal.
- .FOCKDIAGONAL
- Default for CAS calculations.
Use an approximate orbital Hessian diagonal which only uses Fock
contributions.
- .FOCKONLY
- Activate TRACI option (default : program decides).
Modified TRACI option where all orbitals, also active orbitals, are
transformed to Fock type orbitals in each iteration.
- .FROZEN CORE ORBITALS
- READ (LUINP,*) (NFRO(I),I=1,NSYM)
Frozen orbitals : Number of inactive (doubly occupied) orbitals to be frozen
in each symmetry (the first NFRO(I) in symmetry I) in MCSCF.
Active orbitals and specific inactive orbitals can be frozen with ".FREEZE"
under *ORBITAL INPUT.
Frozen orbitals in SCF are specified in the *SCF INPUT input module.
- .MAX CI
- READ (LUINP,*) MAXCIT
maximum number of CI iterations before MCSCF (default = 3).
- .MAX MACRO ITERATIONS
- READ (LUINP,*) MAXMAC
maximum number of macro iterations in MCSCF optimization (default = 15).
- .MAX MICRO ITERATIONS
- READ (LUINP,*) MAXJT
maximum number of micro iterations per macro iteration in MCSCF optimization
(default = 24).
- .MAXABS
- READ (LUINP,*) MAXABS
maximum number of macro iterations with
absorption (default = 3).
- .MAXAPM
- READ (LUINP,*) MAXAPM
maximum number orbital absorptions within
a macro iteration
(APM : Absorptions Per Macro iteration; default = 5)
- .NATONLY
- Activate TRACI option (default : program decides).
Modified TRACI option where the inactive and secondary orbitals are not
touched (these two types of orbitals are already natural orbitals).
- .NEO ALWAYS
- Always norm-extended optimization (never switch to Newton-Raphson).
Note: ".NR ALWAYS" and ".CORERELAX"
takes precedence over ".NEO ALWAYS".
- .NO ABSORPTION
- Never orbital absorption (default settings removed)
- .NO ACTIVE-ACTIVE ROTATIONS
- No active-active rotations in RAS optimization.
- .NOTRACI
- Disable TRACI option (default : program decides).
- .NR ALWAYS
- Always Newton-Raphson optimization (never NEO optimization).
Note: ".NR ALWAYS" takes precedence over
".NEO ALWAYS".
- .OLSEN
- Use Jeppe Olsen's generalization of the Davidson
algorithm.
- .OPTIMAL ORBITAL TRIAL VECTORS
- Generate "optimal" orbital trial
vectors [21].
- .ORB_TRIAL VECTORS
- Use also orbital trial vectors as start vectors for auxiliary roots
in each macro iteration (CI trial vectors are always generated).
- .PHPRESIDUAL
- Select configurations for PHP matrix based on largest residual
rather than lowest diagonal elements.
- .SIMULTANEOUS ROOTS
- Default : NROOTS = ISTATE, LROOTS = NROOTS
READ (LUINP,*) NROOTS, LROOTS
NROOTS = Number of simultaneous roots in NEO
LROOTS = Number of simultaneous roots in NEO at start
- .STATE
- Default = 1
READ (LUINP,*) ISTATE
Index of MCSCF Hessian at convergence (1 for
lowest state, 2 for first
excited state, etc. within the spatial symmetry and
spin symmetry
specified under *CONFIGURATION INPUT).
- .SYM CHECK
- Default: ICHECK = 2 when NROOTS
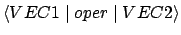 1, else ICHECK = -1.
1, else ICHECK = -1.
READ (LUINP,*) ICHECK
Check symmetry of the LROOTS start CI-vectors and remove those which
have wrong symmetry (e.g. vectors of delta symmetry in a sigma
symmetry calculation).
ICHECK < 0 : No symmetry check.
ICHECK = 1 : Remove those vectors which do not have the same
symmetry as the ISTATE vector, reassign ISTATE.
ICHECK = 2 : Remove those vectors which do not have the same
symmetry as the lowest state vector before selecting
the ISTATE vector.
other values: check symmetry, do not remove any CI vectors.
The ".SIMULTANEOUS ROOTS" input will automatically be
updated if CI vectors are removed.
- .THRCGR
- READ (LUINP,*) THRCGR
Threshold for print of CI gradient. Default is 0.1D0.
- .THRESH
- Default = 1.0D-05
READ (LUINP,*) THRMC
Convergence threshold for energy gradient in MCSCF optimization.
The convergence of the energy will be approximately the square of this
number.
- .TRACI
- Activate TRACI option (default : program decides).
Active orbitals are transformed to natural orbitals and the CI-vectors
are counter-rotated such that the CI states do not change. The
inactive and secondary orbitals are transformed to Fock type orbitals
(corresponding to canonical orbitals for closed shell Hartree-Fock).
For RAS wave functions the active orbitals are only transformed
within their own class (RAS1, RAS2, or RAS3) as the wave function is
not invariant to orbital rotations between the classes. For RAS, the
orbitals are thus not true natural orbitals, the density matrix is
only block diagonalized. Use ".IPRCNO" (see
p. ![[*]](crossref.png) ) to control output from this
transformation.
) to control output from this
transformation.
Comments:
COREHOLE: Single core-hole calculations are
performed as RAS calculations where the opened core orbital is in
the RAS1 space. The RAS1 space must therefore contain one and
only one orbital when the COREHOLE option is used, and the
occupation must be restricted to be exactly one electron. The
orbital identified as the core orbital must be either inactive or
the one RAS1 orbital, if it is inactive it will switch places with
the one RAS1 orbital. The core orbital (now in RAS1) will be
frozen in the following optimization. After this calculation has
converged, the CORERELAX option may be added and the core orbital
will be relaxed. When CORERELAX is
specified it is assumed that the calculation was preceded by a
frozen core calculation, and that the
orbital has already been moved to the RAS1 space. Default
corresponds to the main peak, shake-up energies may be obtained by
specifying ".STATE" larger than one. Absorption is
very beneficial in core hole calculations because of the large
orbital relaxation following the opening of the core hole.
ABSORPTION: Absorption level 1 includes occupied - occupied rotations
only (including active-active rotations); level 2 adds inactive -
secondary rotations and only active - secondary rotations are excluded
at this level; and finally level 3 includes all non-redundant rotation
for the frozen CI vector. Levels 1 and 2 require the same integral
transformation (because the inactive - secondary rotations are
performed using the P-supermatrix integrals) and level 1 is therefore
usually not used. Level 3 is the normal and full level, but it can be
advantageous to activate level 2 together with level 3 if big
inactive-active or occupied-occupied rotations are expected.
ORB_TRIAL: Orbital trialas
start vectors can be used for
excited states and other calculations with more than one simultaneous
roots. The orbital start trial vectors are based on the eigenvectors of
the NEO matrix in the previous macro iterations. However, they are
probably not cost-effective for multiconfiguration calculations where
optimal orbital trial vectors are
used and they are therefore not used
by default.
SYM CHECK: The symmetry check is performed on the matrix element
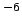 , where "oper" is
the CI-diagonal.
It is recommended and the default to use ".SYM CHECK"
for excited states, including
CI vectors of undesired symmetries is a waste of CPU time.
, where "oper" is
the CI-diagonal.
It is recommended and the default to use ".SYM CHECK"
for excited states, including
CI vectors of undesired symmetries is a waste of CPU time.
DETERMINANTS: The kernels of the CI sigma routines and density matrix
routines are always performed in determinant
basis. However, this
keyword specifies that the external representation is Slater
determinants as well. The default is that the external representation
is in CSF basis as
described in chapter 8 of MOTECC-90. The external
CSF basis is
generally to be preferred to be sure that the converged
state(s) have pure and correct spin symmetry, and
to save disk space.
It is recommended to specify ".PLUS COMBINATIONS" under
"*CI VECTOR" for
calculations on singlet states with
determinants,
in particular for
excited singlet states which often have lower lying triplet states.
Next: *ORBITAL INPUT
Up: Main input groups in
Previous: *NEVPT2 INPUT
Contents
Index
Dalton Manual - Release 1.2.1