Press this badge for direct link to my Researcher ID
My career is centered on the understanding of electron density, and its interplay with the energy and properties of molecules and solids, both from calculations and X-Rays.
Following this principle, I have developed three main axes of research (and several collateral research/hobbies - many due to collaboration with good frieds).
On the one hand, I work on the development, implementation and application of topological tools to analyze the chemical bond in real space, as opposed to Hilbert space - very common in molecules, or reciprocal space, very common in solids.
And I make this "solid" remark because many of my studies are focused in periodic conditions, yielding my second axis of research. Unlike in molecules, we can fine-tune interatomic distances in solids, which constructs the scenario for a wornderful playground. Indeed, high pressure experiments yield amazing new chemistries that still need to be thoroughly explored. Rigorous techniques are needed to understand how the properties of these new states of matter are related to the microscopic structure.
Finally, we are recently interested in understanding and disentangling errors in the triad density-energy-properties, resorting to experimental wavefunctions and trying to estimate error bars which facilitate collaborations with experimentalists...and it is being a lot of fun!
With the increase in computing power, bigger and more complex systems are available for quantum mechanical calculations. Due to its computational cost, DFT is the method of choice in the most majority of cases.
Hence controlling the errors introduced in the simulation becomes a central issue. This project is devoted to the development of mathematical and physical errors expected in a DFT simulation:
The model error, that is the difference between the value of the observable for the exact solution of the Schrödinger equation and the value obtained with the chosen approximate model to solve the equation (e.g. DFT with a given functional).
Based on the assumption that the known Delocalization Error of DFT is the main error source in solid state calculations, we have considered the pure HF and pure DFT(LDA) results as providing the extremes for any other DFT functional, effectively providing an error bar that brings information on how much a specific property of a specific system is affected by the choice of the DFT functional.
The discretization error, that is the difference between the value of the observable for the approximate model and the value obtained with the chosen discretization (eg. basis set) of the approximate model.
We are coupling the error bars from the model with those dealing with the discretization errors both for planewaves or localized basis. These errors are evaluated considering the residual and evaluate its norms accordingly. The evaluation of optimal constants is here of prime importance to fairly represent this part of the error.
The algorithmic error. The discretized model is solved numerically by various iterative algorithms (fixed point for nonlinearities, gradient types for the linear part), which are stopped when a given difference in energy threshold is reached.
We aim at understanding and disentangling convergence in the electron density and functional errors at the model level.
This will enable us to construct density bases which can be fed to experimental techniques (e.g. Hirshfeld atom refinement - HAR). This should be a complete revolution for the X-Ray community, which needs to overcome the simplicity of the multipolar model while lowering the cost of HAR.
We must take into account that many of the ideas related
to the chemical bond constitute a collection of vague and phenomenological
concepts that were originated previously to the birth of quantum mechanics.
In spite of their lack of rigor, their connection to experimental results stands out.
It has been one of the main aims of quantum chemistry within the last decades to provide
a rigorous definition of such useful concepts.
The first step to understand the geometrical and electronic structures in complex
systems clearly requires recovering the concepts of "localization" and "electronic pair"
for these compounds. Hence the Electron Localization Function is one of our aims of study,
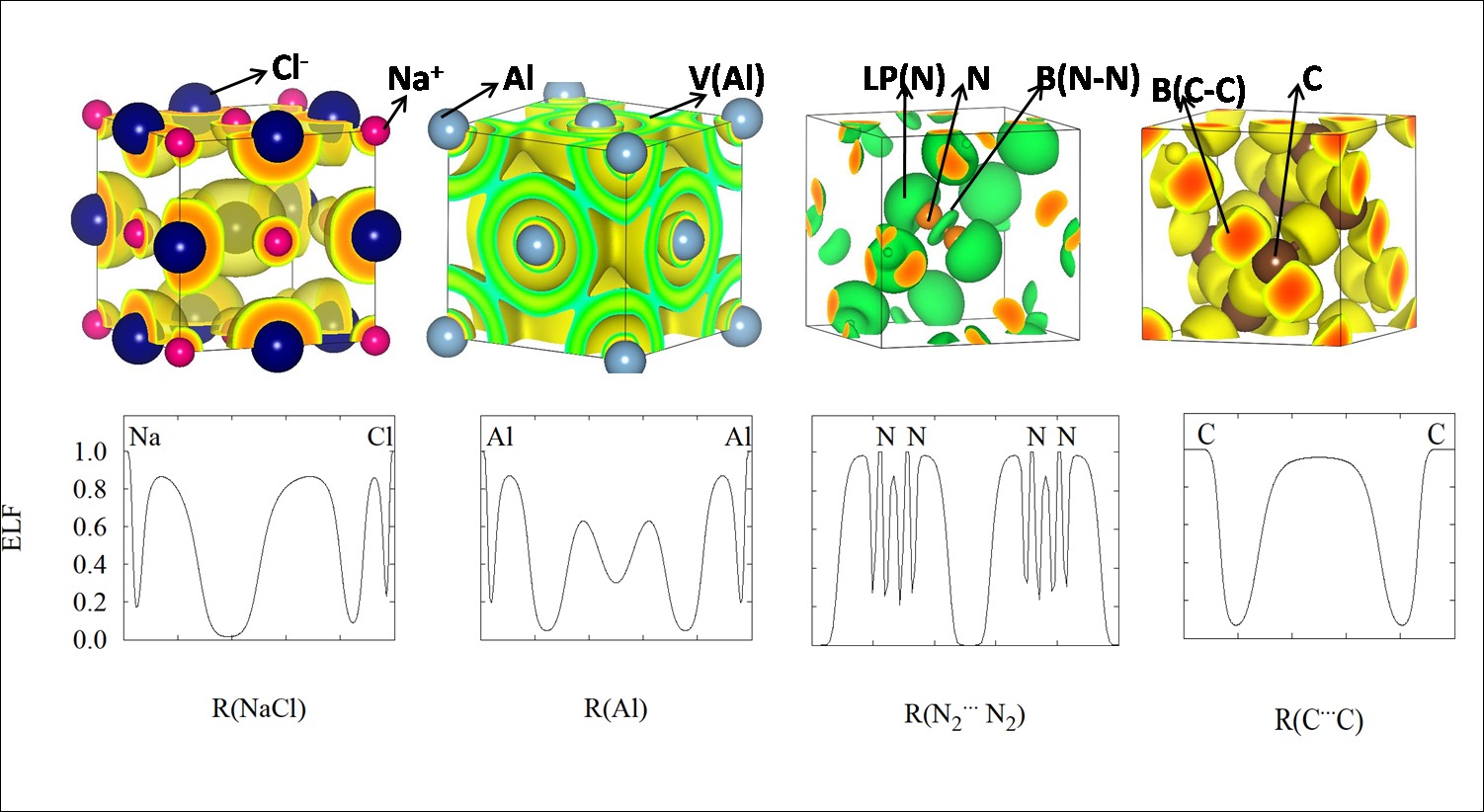
with special focus in periodic systems (Figure 1).
Figure 1. Analysis of bonding patterns in solids thanks to CRITIC
But non covalent interactions might become even more crucial when talking about reactivity,
stability of big systems or catalysis. Hence we also work intensely on tools for revealing
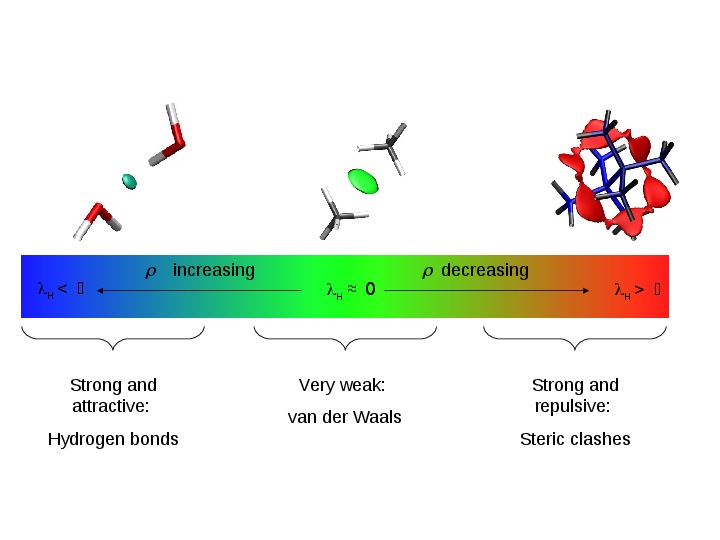
non-covalent interactions in real space, such as the reduced density gradient (Figure 2a).
a)
b)
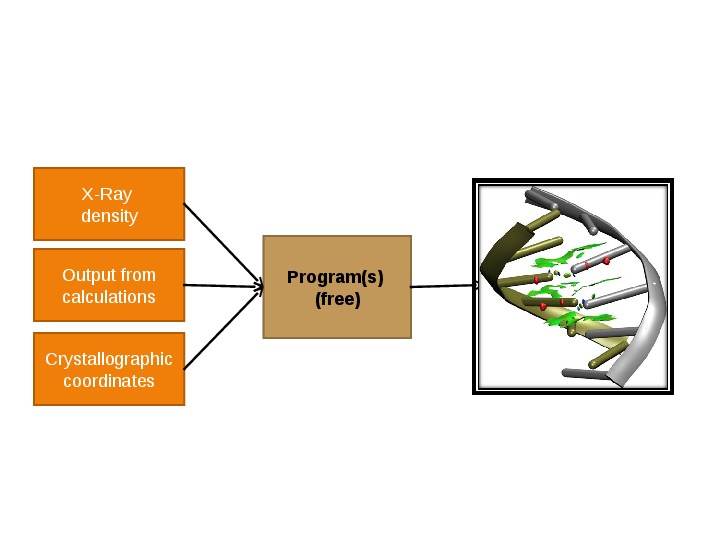
Figure 2. a) Analysis of various non covalent interactions with the reduced density gradient. b) The reduced density gradient can be calculated from theoretical (optimized or promolecular) as well as from experimental densities.
Within this approach, we carry work along the three axis of theoretical chemistry: development,
implementation (Figure 2b) and application.
The use of high pressure to explore interatomic interactions which are
responsible for all physicochemical properties of condensed matter is based
on the simple observation that these interactions, whatever their description,
explicitly depend upon interatomic distances, so that the most direct way to
explore interatomic potentials is to vary interatomic distances.
This is most naturally achieved by applying pressure.
This way, high pressure becomes a wonderful playground to test chemical
interactions and their change.
It is even possible to go one step further and use computations which
have been validated against precise high pressure data to solve problems
which cannot be solved by experiment alone.
Understanding new chemistries that appear under pressure is one of our basic aims and
main areas of application of our topological developments. One such example are insulating metals,
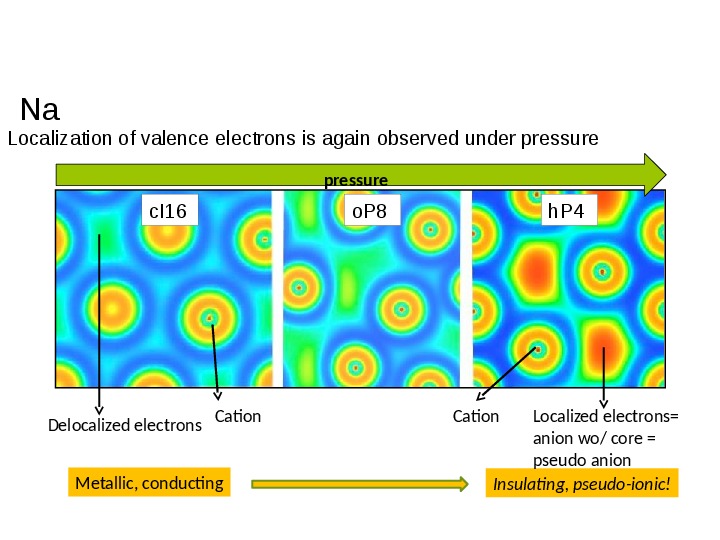
a "pseudo-ionic" electronic structure that appears when alkali metals are submitted to very high pressures (Figure 3).
Figure 3. Evolution of electron localization in sodium under pressure and its relationship with macroscopic properties. Phase hP4 is insulating.
Within this research axis, we have established and important collaboration network with
physicists in the experimental and theoretical fields, trying to unveil the relationships
that determine stability as well as transformations under pressure, and seeking to
construct an understanding of electronic structure that merges real and reciprocal space
microscopic insight with macroscopic and measurable properties.